
Abstract
In this study, we have investigated the hypothesis that previously reported beneficial effect of peripheral blood mononuclear cells cultured under angiogenic conditions on cardiovascular function following ischemia is not limited to EPCs but also to monocytes contained therein. We first purified and analyzed the phenotype and secretome of human and murine blood monocytes cultured under angiogenic conditions (named MDs for monocyte derivatives) and tested their effect in a mouse model of myocardial infarction (MI). FACS analysis of MDs shows that these cells express mature endothelial cell markers and that their proliferative capacity is virtually absent, consistent with their end-differentiated monocytic ontogeny. MDs secreted significant levels of HGF, IGF-1, MCP-1, and sTNFR-1 relative to their monocyte precursors. MDs were unable to form vascular networks in vitro when cultured on matrix coated flasks. Treatment of murine HL-1 cardiomyocyte cell line with MD-conditioned medium reduced their death induced by TNF-α, staurosporine, and oxidative stress, and this effect was dependent upon MD-derived sTNFR-1, HGF, and IGF-1. We further demonstrate that MD secretome promoted endothelial cell proliferation and capacity to form vessels in vitro and this was dependent upon MD-derived MCP-1, HGF, and IGF-1. Echocardiography analysis showed that MD myocardial implantation improved left ventricle fractional shortening of mouse hearts following MI and was associated with reduced myocardial fibrosis and enhancement of angiogenesis. Transplanted MDs and their secretome participate in preserving functional myocardium after ischemic insult and attenuate pathological remodeling.
Keywords
Introduction
Heart failure is the leading cause of morbidity and mortality in developed countries. Postinfarction heart failure resulting from ventricular remodeling remains an issue, although early reperfusion therapy has significantly reduced mortality rates. Promoting the myocardium's natural adaptive responses is the goal of many therapeutic angiogenic strategies. Growth of new blood vessels occurs through vasculogenesis (mobilization and homing of bone marrow-derived progenitor cells), arteriogenesis (remodeling of vessels), and angiogenesis (migration and proliferation of preexisting endothelial cells) (9). In adults, following an ischemic event, new blood vessel formation is characterized by migration of bone marrow-derived endothelial progenitor cells (EPCs) to the site of ischemia and/or by stimulation of capillary sprouting at the site of injury (34, 37, 48).
Circulating bone marrow (BM)-derived EPCs were first identified by Asahara et al. as a subset of CD34+ hematopoietic progenitor cells (2). CD34+ cells cultured in presence of growth factors for 7 days on fibronectin-coated flasks differentiated into endothelial-like cells. These cells incorporated acetylated low-density lipoprotein (AcLDL), bound Ulex (UEA-1) lectin, and formed tube-like structures in vitro (2, 35). Cells cultured from unfractioned peripheral blood mononuclear cells (PBMNCs) were also used as a source of EPCs. These cells were variably termed circulating cultured angiogenic cells (CACs) (42) culture-modified mononuclear cells (CMMCs) (18) or EPCs (22). EPCs obtained from unfractioned PBMNC have been shown to specifically express CD34 and to secrete several growth factors, including VEGF, G-CSF, SDF-1, and HGF (42, 49).
It has been reported that endothelial-like cells may differentiate from cells of the monocytic lineage, suggesting a closer relationship between the monocyte/macrophage and the endothelial cell systems than previously supposed (15). Our hypothesis was that previously reported beneficial effect of PBMNCS on cardiovascular function following ischemia is not limited to EPCs but also to monocytes contained therein.
In the present study, we demonstrate that a purified monocyte population cultured under angiogenic conditions (hereafter called MDs for monocyte derivatives) secrete high levels of antiapoptotic, anti-inflammatory, and proangiogenic factors, which we show play a material role in protecting cardiomyocytes from apoptosis, stimulate angiogenesis, and preserve cardiac function following myocardial infarction (MI).
Materials and Methods
Reagents
Anti-CD31, anti-VEGFR1, and anti-VEGFR2 antibodies used for FACS, ELISA for IGF-1, HGF, sTNFR-1, and MCP-1, as well as HGF and MCP-1 neutralizing antibodies were obtained from R&D systems (Minneapolis, MN, USA). Phospho-Akt, phospho-Bad, and cleaved caspase-3 antibodies used in Western blots were purchased form Cell Signaling Technology (Danvers, MA, USA). IGF-1 neutralizing antibody and DilAcLDL staining were ordered from Cedarlane Laboratories (Toronto, Ontario, Canada). C-met and IGF-1R antibodies were purchased from Abcam (Dresden, Germany). Claycomb medium, endothelial cell growth supplement, Ulex, TNF-α, staurosporine, and cobalt chloride (CoCl2) were supplied by Sigma-Aldrich (St. Louis, MO, USA). Anti-mouse CD106 was purchased from Biolegend. ELISA for cardiac troponin I (cTNI) and Annexin V-PI kit were respectively obtained from Life Diagnostics (Westchester, PA, USA) and Biosource (Caramillo, CA, USA). Anti-CD34, anti-CD45, and streptavidin were obtained from BD Biosciences (Mississauga, Ontario, Canada). VWF antibody was purchased from Fisher Scientific (Ottawa, Ontario, Canada). RPMI was purchased from Hyclone (Logan, UT, USA). EBM-2 medium and single quotes containing bFGF, EGF, VEGF, IGF-1, and ascorbic acid were obtained from Cambrex (Walkersville, MD, USA).
Cell Isolation and Culture
Monocytes and MDs
Mononuclear cells were obtained by Ficoll gradient separation and were enriched in monocytic fraction using the CD14+ RosetteSep kit following the manufacturer protocol (Stemcell Technologies, Vancouver, BC, Canada). Cells were then resuspended either in RPMI or EBM-2 media and seeded in six-well or 24-well plates coated with fibronectin (10 μg/ml). Medium used to culture MDs consisted of EBM-2 supplemented with 1% FBS, 1% Pen/Strep, IGF-1, EGF, FGF, VEGF, ascorbic acid. Monocytes were cultured in RPMI-based medium supplemented with 10% FBS, 1% HEPES, 1% sodium pyruvate, and 1% Pen/Strep. Medium was changed at day 3 following cell plating and was then changed every 2 days. Cells were kept in culture at 37°C in a tissue culture incubator (5% CO2) for no more than 9 days.
HL-1 Cells
The HL-1 cell line derives from tumoral atrial cardiomyocytes from transgenic mice and was a gift from Dr Alvin Shrier (McGill University, Montreal, Canada). These cells have been shown to contract and to retain phenotypic characteristics of the adult cardiomyocytes (12). In addition, HL-1 cells have been widely used to study mechanisms involved in cardiac apoptosis induced by several factors, including oxidative stress, endotoxins, and antihypertensive agents (8, 29, 30, 45, 51). Cells were cultured in Claycomb medium supplemented with 10% fetal bovine serum, 4 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.3 mM ascorbic acid, and 10 μM norepinephrine, at 37°C in a humid atmosphere of 5% CO2/95% air. T75 flasks (used for cell expansion), and six-well plates (used for cell death and proliferation assays) were coated with a mixture of gelatin (0.02%) and fibronectin (25 μg/ml) and the Claycomb medium was renewed daily. Cells were split at a mean density of 15,000/cm2 and cultured till they reached 75% confluence.
Mouse Cardiac Endothelial Cells (CEC)
A cell pellet was obtained from mouse hearts after enzymatic digestion with type II collagenase at 37°C for 2 h. Cells were then washed twice with PBS and CEC were then seeded in T75 flasks in DMEM supplemented with 2 mM glutamine, 1% penicillin/streptomycin mixture, 1 mM sodium pyruvate, 20 mM HEPES, 1% nonessential amino acids, 20% FBS, 150 μg/ml endothelial growth supplement, and 12 U/ml heparin. Cells were kept in a humidified atmosphere of 5% CO2/95% air. A 24-h preplating step and cell sorting were performed to purify the endothelial cell population. Cells that were CD45-, CD106+, and CD31+ were considered as CEC. CEC were used at passage 3 and were passed before reaching 80% confluence.
HUVEC
These cells were obtained from BD Bioscience (Mississauga, Ontario, Canada). They were cultured in DMEM supplemented with 2 mM glutamine, 1% penicillin/streptomycin mixture, 1 mM sodium pyruvate, 20 mM HEPES, 1% nonessential amino acids, and 20% FBS. HUVEC were used at passage 3 and were passed before reaching 80% confluence.
Cell Labeling for FACS Analysis
Cells were resuspended in PBS containing 1% FBS buffer and incubated with either a unique antibody conjugated to APC or a biotin conjugated antibody for 2 h at 4°C. The latter were then incubated with streptavidin PE-Cy7 antibody for 1 h at room temperature. FACS was performed using a FACScalibur analyzer (BD Bioscience).
Protein Array Assay
The cytokine array protocol was used on MD- or monocyte-conditioned media following step by step the manufacturer's instructions (RayBiotech, Norcross, GA, USA). Briefly, arrays were first blocked with a blocking buffer. Conditioned medium (test sample) or FBS (control sample) was used for the assay. A mouse array was used for qualitative evaluation of the expression of 10 proteins: IGF-1, IL-10, MCP-1, MMP-2, PMMP-9, SDF-1, sTNFR-1, TNF-α, TIMP2, and VEGF. The human array consisted of 20 proteins: EGF, GCSF, GM-CSF, HGF, IGF-1, IL-6, IL-10, IL-15, IL-1Ra, MCP-1, MMP-1, MMP-9, SDF-1, sTNFR-1, TGF-β, TIMP-1, TIMP-2, TNF-α, VE-cadherin, and VEGF. Determination of protein expression was performed by analysis of the blot surface by spot densitometry. Arrays were thus scanned at 600 dpi resolution (HP scanner model 1200) and acquired images were then transferred to Scion Image software (version 4.0.2). Blots were discriminated from surrounding background using the thresholding mode. Evaluation of stained areas was performed by blot density and surface measurement.
In Vitro Cell Survival Assay
A total of 500,000 HL-1 cells were plated in six-well plates in low serum medium (2% FBS Claycomb medium) supplemented with MD-conditioned medium. After 48 h, staurosporine (1 μM), TNF-α (20 ng/ml), or CoCl2 (200 μM) were added to some wells. Cell viability was analyzed using Annexin-V-PI cell death detection kit according to the manufacturer's protocol (Life Diagnostics, USA).
Evaluation of Proliferation of MDs, Monocytes, HL-1 Cells, and CEC
Cells were seeded in six-well plates at a density of 200,000 cells per plate in specific culture medium. Cells were trypsinized (1 ml per well) and harvested at the beginning of the experiments (T0) and 2, 5, and 7 days later. Cells were counted using Z2 Beckman Coulter particle counter (Miami, FL, USA) at the indicated time points.
Angiogenesis Assay
Ninety-six-well plates were first coated at 37°C for 2 h with a matrix containing laminin, collagen type IV, heparin sulfate proteoglycans, entactin, and nidogen. Cells (1 × 104) were seeded in each well and were incubated in the presence or absence of MD-conditioned media for 16 h.
In Vitro Angiogenesis Score Quantification
Visual patterns were defined on photos of five random view-fields per well. A numerical score was then assigned to each condition according to the degree of angiogenesis progression described in Table 1. The pattern association criterion was defined by the number and size of polygons formed, capillary thickness, and cell alignment and fusion.
In Vitro Angiogenesis Score Determination

Visual patterns were defined on photos at 40× magnification of five random view-fields per well. The score corresponds to the degree of angiogenesis progression. The pattern association criterion was defined by the number and size of polygons formed, capillary thickness, and cells alignment and fusion.
Western Blot
HL-1 cells were harvested and resuspended in a lysis buffer. Protein (40 μg) was fractionated in a 12% polyacrylamide SDS gel and then transferred onto a polyvinylidene fluoride (PVDF) membrane (Bio-Rad, Ontario, Canada). The membrane was then incubated with a primary antibody (anti-mouse phospho-Akt or anti-mouse phospho-Bad) overnight at 4°C. The blots were then incubated for 1 h with their respective secondary antibody at room temperature. Blots were visualized using chemioluminescence kit (Millipore, Montreal, Canada).
RT-PCR for CCR-2
CCR-2 expression was evaluated by RT-PCR on RNA obtained from HL-1 and endothelial cells using primers purchased from R&D Systems (Montreal, Canada) and following the manufacturer protocol.
Animal Models
All experiments were carried out in agreement with local guidelines for the care and use of laboratory animals and in accordance to the guidelines of the McGill University animal care authority. Investigations conform to the US National Institutes of Health Guide for the Care and Use of Laboratory Animals. In vivo experiments were performed using 10 mice per group. Results presented in this study include mice with the same heart rate and with similar infarct sizes. Cells were obtained from C57Bl/6 mouse peripheral blood and injected into C57BL/6 female mice at 10–14 weeks of age used as recipients for MDs and monocytes. Myocardial infarction was induced by ligation of left anterior descending coronary artery. Cells were resuspended in PBS for injection at a concentration of 15,000 cells/μl of PBS. A total of 300,000 cells or the equivalent PBS volume was injected 10 min after ligation per mouse with a nontraumatic 32-gauge needle (Hamilton, USA) into two different ischemic sites.
Criteria of Inclusion in the Infarct Study
After the onset of an MI, cTnI levels increase substantially and are measurable in serum within 4–6 h, with peak concentrations reached in approximately 12–24 h after infarction. In our experiments, the infarct size was estimated by determination of cTnI in mouse plasma collected 24 h post-MI, as previously described (14, 38). Infarct size was estimated by ELISA for cTnI following the manufacturer's protocol. Analysis of cardiac echograms and study of myocardial histology were performed on mice with a cTnI concentration of 28 ± 6 ng/ml.
Echocardiography
Echocardiography was performed as described previously with some modifications (1), 1 month after the induction of MI. Two-dimensional guided M-mode echocardiography was performed under light isoflurane anesthesia (0.75–1% isoflurane, 1 L/min O2) using a Visual Sonic VEVO 770 ultrasound machine and a RMV™ 707B High Frame Rate Scanhead with a center frequency of 30 MHz. The percentages of isoflurane and O2 flow were adjusted to obtain the fastest heart rate (HR) possible. Left ventricle (LV) end-diastolic and end-systolic internal diameter (LVIDd and LVIDs), end-diastolic interventricular and LV posterior wall thickness (IVSd and LVPWd), and fractional shortening (FS) were determined as described previously (1, 24). Mice with HR between 435 and 485 bpm were included in the study.
Heart Dissection and Histological Analysis of Infarct Area
Mice were anesthetized with isofurane and a cut was made in the peritoneum to localize the posterior vena cava (PVC). Eight hundred microliters of 0.1 M KCl was injected through the PVC to stop the heart in diastole. An incision was performed over the left thoracic area. Muscles over the ribs were delicately separated to visualize the heart. The pericardium was carefully opened and the heart was removed. Harvested hearts were then injected with 0.5 ml of PBS/1% heparin via the superior vena cava. For histological analysis, hearts were fixed in 10% formalin and mounted in paraffin. Paraffin sections were stained for H&E or Sirius red and processed for VWF immunohistology. The latter was done at the histology facility of Immunology and Cancer Research Institute of Montreal University (Quebec, Canada). To evaluate the surface of Sirius red and VWF staining, photos of heart sections were acquired using a Leica DM-LB2 microscope with a 20× objective. Images were then transferred to Scion Image software (version 4.0.2). Tunel labeling was performed following step by step the manufacturer's protocol. In order to avoid false positive Tunel results, heart sections were treated with diethyl pyrocarbonate as previously reported (47). Objects of interest (Sirius red or VWF staining surfaces and labeled nuclei) were discriminated from surrounding background using the thresholding mode. In this mode, pixels equal to or greater than threshold level are displayed in black and all other pixels are displayed in white (background). Evaluation of stained areas was performed by surface measurement.
Statistical Analysis
Data are presented as means ± SEM. Differences among groups were statistically analyzed using analysis of variance. Tests were followed up by Bonferroni correction within groups. Statistical analyses were performed using GraphPad software. A value of p < 0.05 was considered significant.
Results
MDs and Mature Endothelial Cells Have Common Molecular Markers
MDs are purified monocytes obtained from mouse or human peripheral blood and cultured under angiogenic conditions (EBM-2 medium supplemented with IGF-1, EGF, VEGF, and FGF on fibronectin-coated flasks). Cells were labeled with DilAcLDL, Ulex, or with CD14, CD31, CD34, CD45, VEGFR1, VEGFR2, and VWF antibodies. FACS analysis showed that MDs were positive for CD14, CD31, CD45, VWF, VEGFR-1, VEGFR-2, lectin, and DilAcLDL, and negative for CD34 (Fig. 1A-C). In contrast, endothelial cells were negative for CD14, CD34, and CD45 and positive for CD31, VWF, VEGFR-1, VEGFR-2, lectin, and DilAcLDL (Fig. 1D–F). Analysis of four different human samples and 50 mouse samples indicated that over 97% of mouse as well as human MDs were positive for CD14 after purification. These results demonstrate that MDs did not differentiate into endothelial cells but maintained their monocytic phenotype. In addition, human and mouse MDs cultured in EBM-2 medium up to 2 weeks did not proliferate and were unable to form vascular cords in vitro when cultured on matrix coated flasks (Fig. 1G).
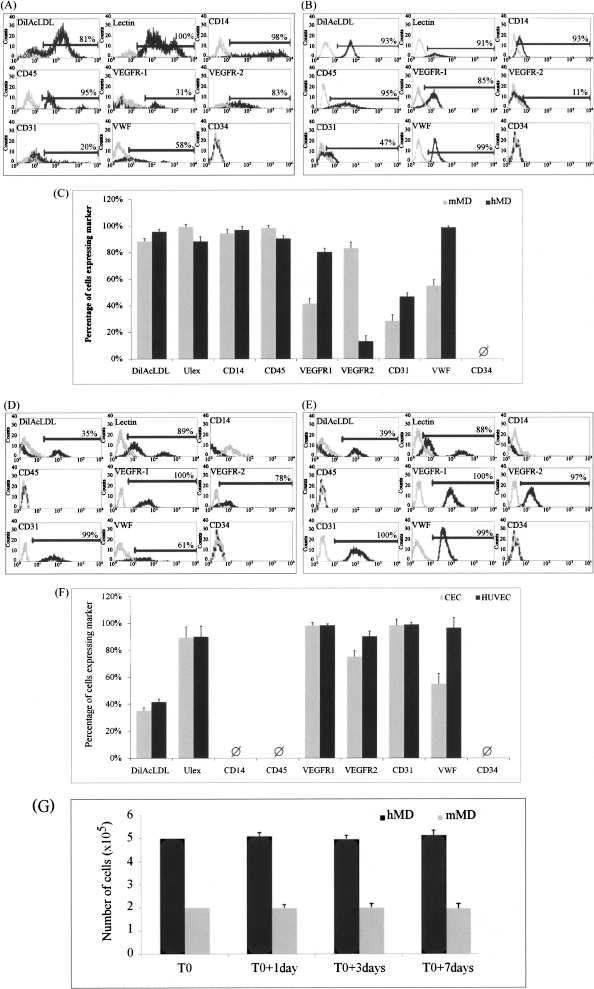
FACS analysis of mouse versus human MDs and endothelial cells. Mouse and human MD samples were analyzed for indicated markers by FACS. The plots show the intensity of staining of mouse (A) and human (B) MDs with specific (black lines) and isotype control (gray lines) antibodies. (C) Histogram representation of mouse (gray bars) and human (black bars) MDs phenotyping analysis. The plots in (D) and (E) show the intensity of staining of CEC and HUVEC with specific (black lines) and isotype control (gray lines) antibodies, respectively. (F) Histogram representation of CEC (gray bars) and HUVEC (black bars) phenotyping analysis. (G) MD proliferation assay. Human MDs (hMD; 5 × 105) and 2 × 105 mouse MDs (mMD) were plated in six-well plates and counted at the indicated time points. MDs are purified monocytes obtained from mouse or human peripheral blood and cultured under angiogenic conditions (EBM-2 medium supplemented with IGF-1, EGF, VEGF, and FGF on fibronectin-coated flasks). MDs cultured for 6–7 days resulted in spindle-shape morphology and their proliferative capacity was virtually absent, consistent with their end-differentiated monocyte ontogeny. Ø indicates that no marker expression was detected. Results are reported as mean ± SEM (n = 3).
MDs Release High Levels of sTNFR-1, IGF-1, HGF, and MCP-1
MDs were cultured in EBM-2 supplemented with FBS, IGF-1, VEGF, EGF, and bFGF. Unmanipulated monocytes were cultured in RPMI-1640 supplemented with FBS only. The MD medium was then removed and replaced by EBM-2 supplemented with FBS only. After 48 h of culture, conditioned medium was harvested and protein expression analyzed using protein arrays. Analysis of mouse MD secretome showed high levels of IGF-1, MCP-1, and sTNFR-1 (Fig. 2A and B). Spot densitometry analysis of human MD secretome revealed high amounts of HGF, MCP-1, and sTNFR-1, whereas the level of IGF-1 was low (Fig. 2C and D). ELISAs were performed on mouse and human MD secretome to quantify the expression of IGF-1, MCP-1, HGF, and sTNFR-1 (Table 2). Mouse MDs secreted on average 25-fold more sTNFR-1 than monocytes whereas the quantity of sTNFR-1 secreted by human MDs was on average fivefold higher than monocytes. MCP-1 was highly secreted by mouse and human MDs as well as mouse and human monocytes. In addition, human MDs secreted 14-fold more HGF than monocytes. Mouse MDs secreted high amounts of IGF-1 whereas monocytes secreted low concentration of this protein. In contrast, another series of ELISA demonstrated that mouse MDs secreted low levels of HGF whereas human MDs released very low amounts of IGF-1. Of note, we tested various FBS concentrations and our results indicate that FBS did not modulate levels of these proteins in MD-conditioned medium (data not shown).

Analysis of mouse and human MD secretome. (A, C) Representative images of protein array membrane probed with conditioned media either from mouse and human MDs or from monocytes. (B) Histogram representation of spot densitometry analysis of protein arrays performed on mouse MD (black bars) and monocyte (gray bars) secretomes. (D) Histogram representation of spot densitometry analysis of protein arrays performed on human MD (black bars) and monocyte (gray bars) secretomes. Experiments were performed on three secretomes obtained from three different cell cultures. MD CM, MD-conditioned medium; EBM-2 CPT, EBM-2 culture medium completed with FBS and cytokines; Monocyte CM, monocytes conditioned medium; RPMI-1640, RPMI-1640 medium completed; FBS, fetal bovine serum only. Data are reported as mean ± SEM (n = 3).
ELISA for sTNFR-1, IGF-1, MCP-1, and HGF on MD- and Monocyte-Conditioned Media

Results are normalized to culture medium alone. Data are reported as mean ± SEM (n = 3).
MD-Conditioned Medium Protected HL-1 Cells From Death Induced by TNF-α, Staurosporine, and Oxidative Stress But Had no Effect on Their Proliferation
HL-1 cardiac cells were seeded in six-well plates and cultured for 48 h in Claycomb completed medium (Clay) or in low-serum medium (LSM). Some wells were supplemented with TNF-α, staurosporine, or CoCl2 to induce HL-1 cell death. Mouse or human MD-conditioned media were added then either in presence or absence of HGF, IGF-1, MCP-1, and sTNFR-1 neutralizing antibodies. As shown in Figure 3A, treatment of HL-1 cells with TNF-α reduced by 31.7% the percentage of living cells. Adding mouse MD-conditioned medium to TNF-α supplemented LSM reduced by 20.3% the percentage of dead cells. Interestingly, blocking of IGF-1 and sTNFR-1 with antibodies reduced the percentage of surviving cells cultured in LSM supplemented with mouse MD-conditioned medium and TNF-α by 8.1% and 11.2%, respectively. In contrast, inhibition of MCP-1 had no effect on HL-1 survival rate. In another series of experiments, HL-1 cells were cultured in LSM with either mouse or human MD-conditioned medium and apoptosis was induced by CoCl2 and staurosporine. Figure 3B and C shows protective effect of mouse and human MD-conditioned medium against CoCl2- and staurosporine-induced apoptosis, respectively. Indeed, treatment of HL-1 cells with mouse and human MD-conditioned medium reduced by 21% and 12% the percentage of dead cells in presence of CoCl2. The percentage of living HL-1 treated with staurosporine and mouse and human MD-conditioned medium was also enhanced by 32% and 25%, respectively. Neutralization of IGF-1 and HGF with antibodies reduced the percentage of surviving HL-1 cells incubated with CoCl2 or staurosporine, whereas inhibition of sTNFR-1 and MCP-1 had no effect on their survival when cultured in the same conditions. Incubation of cells in the presence of IGF-1 and sTNFR-1 isotypic antibodies had no effect on their survival (Fig. 3D). Moreover, treatment of HL-1 cells with either mouse or human MD-conditioned media did not alter their doubling time in vitro (data not shown).

Evaluation of the effect of MD-conditioned medium on HL-1 cardiac cell line against induced death. (A) Protective effect of mouse MD-conditioned medium on TNF-α-induced cell death. (B) Protective effect of mouse and human MD-conditioned medium on CoCl2-induced cell death. (C) Protective effect of mouse and human MD-conditioned medium on staurosporine-induced cell death. (D) Effect of MCP-1, HGF, IGF-1, and sTNFR-1 blocking antibodies on survival of HL-1 cells treated with CoCl2 (i), TNF-α (ii), and staurosporine (iii) without MD-conditioned medium. HL-1 cells were treated with CoCl2, TNF-α, and staurosporine in the presence or absence of HGF, IGF-1, and sTNFR-1 neutralizing antibodies. Our results indicate that treatment of HL-1 cells with neutralizing antibodies only had no effect on their survival in vitro. CTL (Clay), Claycomb medium supplemented with 10% FBS; LSM, low serum medium; mCM, mouse MD-conditioned medium; hCM, human MD-conditioned medium; α-IGF-1, IGF-1 neutralizing antibody; α-HGF, HGF neutralizing antibody; α-MCP-1, MCP-1 neutralizing antibody; α-sTNFR-1, sTNFR-1 neutralizing antibody; STR, staurosporine. Data are presented as mean ± SEM (n = 5).*, ψ, κ, ϕ, and ω indicate a significant difference versus bar with identical symbol at p < 0.05.
MD-Conditioned Medium Promoted CEC Proliferation and Angiogenesis via HGF, IGF-1, and MCP-1
CEC obtained from digested mouse hearts were seeded in six-well plates to a density of 1.8 × 105 cells per well. CEC were cultured in low-serum medium for 7 days. Culture medium was changed daily. MD-conditioned medium and MCP-1, sTNFR-1, IGF-1, and HGF neutralizing antibodies were added to the culture medium at the beginning of the experiment (T0) and 5 days later. Results in Figure 4A and B show the effect of human and mouse MD-conditioned media on proliferation of CEC, respectively. Both human and mouse MD-conditioned media enhanced CEC proliferation in vitro. Blocking of HGF, IGF-1, and MCP-1 in the cultured medium using neutralizing antibodies reduced their proliferation, whereas sTNFR-1 inhibition had no effect. These results demonstrate that IGF-1, HGF, and MCP-1 produced by MDs are responsible for the improvement of CEC proliferation in vitro.

Evaluation of the effect of MD-conditioned medium on CEC proliferation and angiogenesis in vitro. (A, B) Growth curve of CEC cultured in LSM in the presence or absence of human or mouse MD-conditioned media. CEC were cultured in low-serum medium (LSM; dark blue line) only or in LSM supplemented with human or mouse MD-conditioned media (hCM or mCM; red line). Some cells were cultured in LSM and MD-conditioned media supplemented with neutralizing antibodies against MCP-1 (α-MCP-1; light blue), sTNFR-1 (α-sTNFR-1; green), HGF (α-HGF; purple), or IGF-1 (α-IGF-1; purple). CEC were counted at the beginning of the experiment (T0) and 2, 5, and 7 days later. Arrows in (A) and (B) indicate time of administration of MD-conditioned media and neutralizing antibodies. (C) Histogram representation of in vitro angiogenesis score of CEC cultured in endothelial cell medium (ECM) in the presence or absence of human (hCM)- or mouse (mCM)-conditioned media. Representative photos of the angiogenesis assay are shown in (D). Average scores for each condition are reported in the top left corner of each photo. Arrows in (D) show alignment of CEC to fuse and form small network.
In a second series of experiments, CEC were seeded in 96-well plates coated with a solid matrix made of basement proteins. CEC were cultured either in endothelial cell medium alone or supplemented with human or mouse MD-conditioned medium. HUVEC were used as an experimental control in this assay. Results in Figure 4C show the in vitro angiogenesis score assessed in each condition according to the degree of cell fusion and network formation. Culture of CEC in angiogenic medium supplemented with human or mouse MD-conditioned media enhanced the angiogenesis score 1.25- and 1.4 fold, respectively, in comparison with CEC cultured in ECM without MD-conditioned media. Representative images of the assay for each condition are depicted in Figure 4D. The latter shows the pattern of the networks and cells defined by the number and size of polygons formed, capillary thickness, and cell alignment and fusion.
MD Implantation Reduced Fibrosis, Promoted Angiogenesis, Reduced Cardiac Cell Apoptosis, and Improved Heart Function Following MI
MI was induced by coronary artery ligation in C57Bl/6 mice and mouse MDs or unmanipulated primary monocytes were injected 10 min after induction of ischemia. It is now well established that evaluation of cTnI plasma levels constitutes a reliable technique to estimate infract size (14, 38). Infarct size was thus estimated by determination of cardiac troponin I (cTnI) in mouse plasma collected 24 h post-MI. Mice with cTnI plasma concentrations between 22 and 34 ng/ml were selected for the in vivo study (Fig. 5A). Echocardiography was performed under isoflurane anesthesia 1 month after the induction of MI plus or minus cell injections. C57Bl/6 mice are very sensitive to anesthesia and have tendency to develop bradycardia. Therefore, because HR might affect the size of the LV chamber and its contractility, only mice with HR between 425 and 485 bpm were included in the study. MI induced a 52.4% decrease in FS and injection of MDs into the ischemic myocardium reduced the MI-induced decrease in FS by 46% (Fig. 5B). Mice injected with MDs presented a 14% higher FS than mice injected with undifferentiated monocytes. Injection of MDs also tended to reduce the LVIDs and thinning of the posterior wall in LVPWd, as shown in Figure 5C. In addition, fibrosis evaluated by Sirius red stain labeling was reduced by 3.8-fold (Fig. 5D). Angiogenesis was improved by 1.3-fold mainly within the scar zone (Fig. 5E). Hematoxylin and eosin staining was also performed to evaluate cell infiltration and results show that, as expected, there were no infiltrating immune cell clusters 1 month post-MI (Fig. 5F). Estimation of apoptosis within mouse hearts indicated that on average 11% of cardiomyocytes were apoptotic between 4 h and 3 days following MI and that MDs reduced mortality rate by 2.1-fold (Fig. 5G and H).
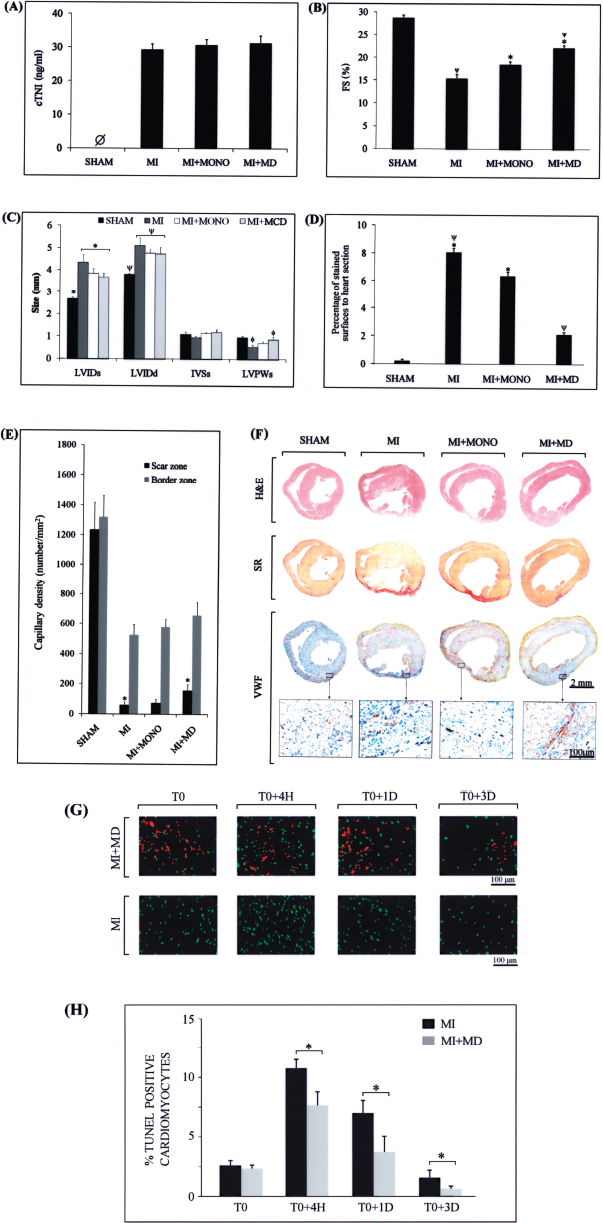
Evaluation of the effect of MD injection into heart following MI. (A) Plasma levels of cardiac troponin I in mice 24 h postsurgery in mice with myocardial infarction (MI) or in sham-operated mice. (B) Left ventricle fractional shortening (FS) and (C) left ventricle (LV) end-diastolic and end-systolic internal diameter (LVIDd and LVIDs), end-sytolic interventricular and LV posterior wall thickness (IVSs and LVPWs) in sham mice and mice with MI with or without injection with monocytes (MONO) or MDs, 1 month following the surgery. (D) Histogram representation of Sirius red-stained surface to whole heart section surface in sham, MI, MI+MONO, and MI+MD mouse groups. (E) Capillary density count within scar (black bars) and border (gray bars) zones. (F) Representative images of heart cross sections stained for hematoxylin and eosin (H&E), Sirius red, and von Willebrand factor (VWF). (G) Representative images of heart cross sections stained for Tunnel (green) and showing PKH26-labeled MDs (red) at the time of injection (T0) and 4 h, 1 day, and 3 days later. (H) Percentage of Tunel-positive cardiomyocytes in mice following myocardial infarction in presence or absence of MDs at the indicated time points. Data are presented as mean ± SEM (n = 5). *, ϕ, and ψ indicate a significant difference versus bar with identical symbol at p < 0.05.
MD-Derived MCP-1, HGF, and IGF-1 Mediate Their Effect Through CCR-2, C-met/Akt, and IGF-1R/Akt Pathways, Respectively
To validate signaling pathways of HGF, IGF-1, and MCP-1, Western blots and RT-PCR were performed on proteins and RNA extracted from CEC, HUVEC, and HL-1 cells. Results in Figure 6A and B indicate that CCR-2 (MCP-1 receptor) was expressed by CEC but not by HL-1 cells. In addition, we demonstrated that c-met (HGF receptor) and IGF-1R (IGF-1 receptor) were expressed on HL-1, HUVEC, and mouse primary CEC (Fig. 6C and D). In addition, treatment of HL-1 cells with TNF-α, CoCl2, or staurosporine enhanced phospho-Bad protein expression while MD-conditioned medium induced Akt phophorylation (Fig. 6E).
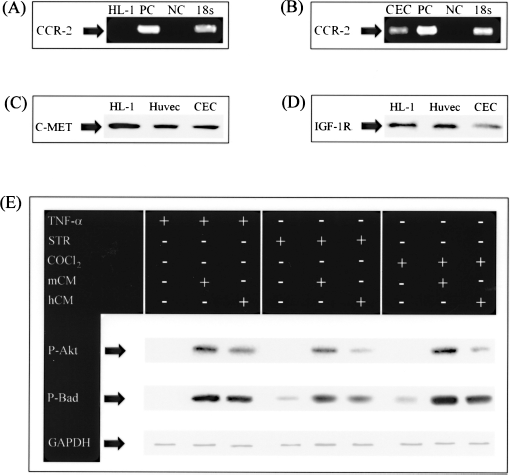
Evaluation of CCR-2, c-met, IGF-1R, phospho-Akt, and phospho-Bad. CCR-2 was evaluated on HL-1 cells (A) and CEC (B) by RT-PCR. Western blots (WBs) were also performed on proteins extracted from HL-1 cells, HUVEC, and CEC for c-met (C) and IGF-1R (D). Proteins were extracted from HL-1 cells treated with staurosporine (STR), cobalt chloride (CoCl2), and TNF-α in the presence of either mouse MD-conditioned medium (mCM) or human MD-conditioned medium (hCM). WBs were performed for quantification of phospho-Akt (P-Akt) and phospho-Bad (P-Bad) (E). PC, positive control; NC, negative control; 18s, internal control.
Discussion
Unfractioned PBMNCs, as routinely collected in humans by a leukapheresis procedure, contain lymphoid cells, monocytes (typically in a 4:1 ratio) as well as CD34+ cells (typically ~1.2% of cells) (16). When this polycellular material is placed in culture with commercially available “endothelial” growth factors, early output cells adopting a partial endothelial phenotype (early EPCs) are obtained. In previous studies, EPCs thus obtained were identified using various endothelial characteristics such as AcLDL uptake, Ulex binding, and CD31, CD34, CD45, CD133, e-NOS, VEGFR1, VEGFR2, and VWF expression (2, 7, 25, 33). However, it was later recognized that these early EPC cultures may contain monocytes (42). We performed our experiments using purified monocytic fraction prior to culture modification to verify whether monocytes could exert regenerative properties. We have referred to the output of this process as monocyte derivatives (MDs) in distinction to culture-modified unfractioned mononuclear cells, which also incorporates lymphoid and CD34+ cells in the mix (23, 42, 49). We compared the molecular phenotype of MDs to mouse primary CEC, HUVEC, EPCs, and monocytes (Figs. 1 and 2, Table 3). We found a phenotypic overlap between endothelial cells and monocytes derived from human and mouse peripheral blood and we demonstrated that these markers cannot be used to distinguish specifically between endothelial cells and monocytes cultured under angiogenic growth conditions.
Comparative Analysis of MD, Endothelial Cell, EPC, and Monocyte Molecular Phenotypes

+++: high level of expression; ++: medium level of expression; +: low level of expression; -: no expression, and ±: have been reported by some research groups and refuted by others [(50)*, (33)†].
Results summarized in Table 3 show that CD14 was expressed by mouse and human MDs as well as by monocytes cultured in RPMI-1640 and has also been reported to be expressed by “cultured circulating EPCs” (27, 28). AcLDL uptake, lectin binding, and expression of CD31, VEGFR2, and VWF markers seem to be characteristic of MDs, endothelial cells, EPCs, and undifferentiated monocytes. The CD45 hematopoietic marker was predictably not detected on endothelial cells. We have shown as well that MDs do not proliferate and were unable to form vascular structure in vitro. Previous publications reported that unfractioned mononuclear cells cultured with angiogenic medium express endothelial markers and secrete cytokines and/or growth factors (23, 42, 49). Our findings are consistent with previous data from Rehman et al. (42), who reported that mononuclear cells isolated from peripheral blood and cultured in endothelial basal medium supplemented with IGF-1, EGF, VEGF, and FGF on fibronectin-coated tissue culture flasks express some monocyte/macrophage markers and secrete growth factors. This cell population was referred to as circulating angiogenesis cells (CACs) and was characterized by the expression of the monocyte activation marker CD11c, the panleukocyte marker CD45, and the early macrophage differentiation marker CD163.
However, there is a methodological and technical difference between previously published data and information provided in this article. Indeed, our experiments were performed using a starting purified population of peripheral blood monocytes cultured under angiogenic conditions that we showed express monocytic as well as endothelial cell markers. According to the study from Rehman et al. (42), CACs were obtained from nonpurified peripheral blood mononuclear cells. CACs consist primarily of monocyte/macrophage-derived cells but they also include a small population of true stem/progenitor cells and endothelial cells. Thus, their results do not exclude the role of these endothelial or progenitor cells in their possible interaction with monocytes. In addition, we provide in vitro and in vivo quantification of the antiapoptotic, angiogenic, and anti-inflammatory effects of secreted cytokines and growth factors. In a recent publication, Hossne et al. investigated in a phase I/IIa clinical trial, the safety and the efficacy of a cell therapy protocol based on multiple intracardiac injections of bone marrow mononuclear cells in patients suffering from refractory angina (21). Their results indicate a correlation between the number of transplanted monocytes and the improvement of cardiac angiogenesis.
Having defined with detail the ontogeny and acquired phenotype of MDs, our study's purpose was to assess the effect of MDs on cardiac and endothelial cell proliferation and survival. We have thus demonstrated that human MDs mediate their antiapoptotic and proangiogenic effects by release of HGF, MCP-1, and sTNFR-1. The beneficial effect of mouse MDs was due to IGF-1, MCP-1, and sTNFR-1. These released proteins reduced cardiac cell apoptosis, enhanced endothelial cell proliferation in vitro, and reduced fibrosis in vivo. These results suggest that IGF-1, sTNFR-1, and MCP-1 released by mouse MDs as well as HGF secreted by human MDs play a key role in the angiogenic and antiapoptotic cardiomyocyte-preserving effects of MDs. The effect of MD-conditioned medium against apoptosis was first evaluated using TNF-α. Indeed, cardiomyocyte death may be caused by TNF-α produced during an inflammatory reaction. TNF-α-induced apoptosis is activated when TNF-α binds to TNFR-1 and thus triggers the release of cytochrome-c by the mitochondria and promotes the executioner caspase-3. In our experiments, sTNFR-1 released by MDs reduced TNF-α-induced cell death by acting as a decoy. Binding of sTNFR-1 to TNF-α reduced its apoptotic activity and enhanced HL-1 cell survival (Fig. 3A). CoCl2 is known to activate hypoxic signaling and is widely used to cause overproduction of reactive oxygen species (ROS) in several cell lines (19, 43, 52). ROS induces apoptosis by the activation of caspase-3-dependent DNA damage. In this case, caspase-3 cleaves the ICAD (inhibitor of CAD), and free CAD (caspase activated DNAse) cleaves DNA (20). MD-conditioned medium protected HL-1 cell death when culture medium was supplemented MD-conditioned media (Fig. 3B). Our experiments demonstrated that IGF-1 produced by mouse MDs and HGF secreted by human MDs were potentially involved in the improvement of HL-1 cell survival when treated with CoCl2. We also evaluated the effect of mouse and human MD-conditioned media on the protection of HL-1 cells from death induced by staurosporine. Our results demonstrate that MD-conditioned media reduced HL-1 cell death induced by staurosporine and that the protective effect was mediated via release of HGF or IGF-1.
To validate signalling pathways of HGF, IGF-1 and MCP-1, we have first demonstrated expression of c-met (HGF receptor), IGF-1R (IGF-1 receptor) and CCR-2 (MCP-1 receptor) on HL-1 and/or mouse primary CEC (Figures 6A, 6B, 6C and 6D). In another series of experiments, we have shown that MD-conditioned medium induced Akt phophorylation while treatment of HL-1 cells with TNF-α, CoCl2 or staurosporine enhanced phospho-Bad protein expression (Figure 6E). These results suggest that proteins released by MDs (e.g: IGF-1 and HGF) enhanced phospho-Akt expression that in turn reduced Bad activity enhancing the HL-1 cell survival rate (32, 36, 39). MCP-1 had no effect on HL-1 cell survival since CCR-2 (MCP-1 receptor) was not expressed. Factors released by MDs enhanced CEC proliferation and promoted angiogenesis in vitro and in vivo (Figs. 4 and 5). This effect seems to be mediated by IGF-1, MCP-1, and HGF. IGF-1 is known to act as a regulator of cell growth via Akt and Erk signaling pathways (4, 6, 10, 17, 41, 46). IGF-1 stimulates synthesis and secretion of extracellular matrix proteins by vascular smooth muscle cells (VSMC) (3) and endothelial cells (5), and it has been shown to be a potent chemoattractant factor for human VSMC (6, 17). Moreover, IGF-1 preserved myocardial structure and improved myocardial function in a swine model of myocardial infarction (31). In contrast, we reported that human MDs secreted high levels of HGF while low concentrations of IGF-1 were detected in their secretome. HGF is widely referred to as a mitogen, morphogen, and angiogenic factor (13, 26). The angiogenic effect of HGF may be achieved by multiple pathways that mediate release of other angiogenic factors such as VEGF (53) or that promote EPC migration by upregulation of inducible nitric oxide synthase (iNOS) expression (40). HGF has been reported to protect cardiomyocytes against oxidative stress via activating the MEK-MAPK pathway (30). Chen et al. have recently shown that HGF gene transfer improved left ventricular ejection fraction and fractional shortening and reduced fibrosis. This approach also increased the capillary density surrounding the infarcted area, suggesting a potential use of HGF in heart healing following MI (11). In addition, HGF has been reported to protect cardiomyocytes against oxidative stress via activating the MEK-MAPK pathway (30). These data suggest that HGF may play a key role in the angiogenic and antiapoptotic effects of human MDs. Interestingly, the identification of CCR-2 in CEC shown here indicates that MCP-1 may not only mediate monocyte infiltration but may also participate in endothelial cell migration and proliferation during angiogenesis.
We have shown here that mouse MD injection into the infarcted myocardium reduced, in a statistically significant manner, the extent of myocardial fibrosis and apoptosis, enhanced the fractional shortening, and tended to reduce the dilatation of the left ventricular chamber in systole (LVIDs) and thinning of the posterior wall in diastole (LVPWd) (Fig. 5). We have also demonstrated that capillary density was greater in mice transplanted with MDs in comparison with the control group transplanted with unmanipulated monocytes (Fig. 5E). MD transplantation may thus reduce left ventricle remodeling after MI mainly via the reduction of cardiomyocyte death and the improvement of angiogenesis. In a recent publication, Ziebart et al. demonstrated that some EPCs obtained from blood mononuclear cells and injected intravenously survive up to 1 month following their transplantation into the infarcted myocardium and that their physical incorporation contributes to the improvement of neovascularization and cardiac function (54). Our results indicate that the majority of MDs do not survive more than 2 weeks in vitro. Because they were injected after 1 week of culture, we presume that these cells mediate their effect shortly after their injection. In order to support this observation in vivo, PKH26-labeled MDs were injected into mouse hearts following MI. Some mice were sacrificed at the time of injection (T0) and 2, 5, and 7 days later. Our results indicate that MDs were visible at the indicated time points, although their number seems to decrease over time (Fig. 5G). Our results do not exclude other cell intrinsic mechanisms by which the cells contribute to improve neovascularization as reported by Ziebart et al. (54).
Several studies demonstrated that recruited monocytes/macrophages regulate angiogenesis in the ischemic tissue (44). Here we have shown that myocardial protection following infarction can be induced in part by growth factors released by MDs. These data strongly support the use of unfractioned PBMNCs containing EPCs as well as monocytes cultured under angiogenic conditions as a source of cells for the treatment of MI.
Footnotes
Acknowledgments
We would like to thank Dr. Alvin Shrier (McGill University, Montreal, Canada) for providing HL-1 cells. This work was supported by Grants 72553 (to J.G.) and 82790 (to E.L.S.) from the Canadian Institutes for Health Research (CIHR), Canadian Stem Cell Network (CSCN), and Fonds de Recherche en Santé du Quebec (FRSQ).