
Abstract
Embryonic stem (ES) cells can be induced to differentiate into motor neurons (MN). Animal models resembling MN degeneration and paralysis observed in familial amyotrophic lateral sclerosis (ALS) have been previously reported. In this work, we aimed to investigate whether transplanted MN could prevent motor deterioration in transgenic rats expressing a mutant form of human superoxide dismutase 1 (hSOD1G93A) associated with inherited ALS. Mouse ES cells were differentiated to neurons that express green fluorescent protein (GFP) under the promoter of the MN-specific gene hb9, as well as molecular markers indicative of MN identity. Cells were grafted into the lumbar spinal cord of adult wild-type (WT) or hSOD1G93A rats at 10 weeks of age, when transgenic animals are presymptomatic. Grafted cells with MN phenotype can survive for at least 1 week in hSOD1G93A animals. To quantitatively evaluate motor performance of WT and transgenic rats, we carried out weekly rotarod tests starting when the animals were 14 weeks old. Sham and grafted WT animals showed no decline in their ability to sustain themselves on the rotating rod. In contrast, sham hSOD1G93A rats decreased in motor performance from week 16 onwards, reaching paralysis by week 19 of age. In grafted transgenic animals, there was a significant improvement in rotarod competence at weeks 16 and 17 when compared to sham hSOD1G93A. However, in the following weeks, transplanted hSOD1G93A rats showed motor deterioration and eventually exhibited paralysis by week 19. At end-stage, we found only a few endogenous MN in sham and grafted hSOD1G93A rats by cresyl violet staining; no choline acetyl transferase-positive nor GFP-positive MN were present in grafted transgenic subjects. In contrast, WT rats analyzed at the same age possessed grafted GFP-positive MN in their spinal cords. These results strongly suggest that the transgenic hSOD1G93A environment is detrimental to grafted MN in the long term.
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease that is characterized by the selective death of motor neurons (MN) in the brain and the spinal cord, leading to generalized weakness, muscle atrophy, and paralysis (6). Currently, there is no effective treatment for this illness, although riluzole can cause a marginal benefit by slowing disease progression (18,27). Approximately 90% of ALS cases are sporadic; in 10% of patients a genetic linkage has been described, and therefore they are classified as familial ALS (FALS). In fact, 20% of FALS cases are caused by dominant mutations in Cu/Zn superoxide dismutase 1 (SOD1) (33), an enzyme that converts the highly oxidizing superoxide to hydrogen peroxide and water. To date, more than 100 mutations in SOD1 are associated with FALS onset (1). One of these mutations, human (h) SOD1G93A has been expressed in transgenic animals (17,20) with pathological findings that have been extensively characterized and closely resemble the MN degeneration and paralysis observed in ALS patients. Thus, these animals are useful models to study potential therapies.
Because of the selective loss of MN, cell replacement therapy is a possible treatment for ALS. Stem cells are self-renewing and can differentiate to specialized phenotypes. Embryonic stem (ES) cells are derived from the inner cell mass of embryos at the blastocyst stage, and are considered to be pluripotent because they can differentiate into cells from the three germinal layers (15,25). It is well established that ES cells have the capacity to generate neuronal cells, and that upon transplantation these neurons cause improvement of neurological deficits in experimental animals (3,7,11,12,23,32,34,37). Induction of ES cells into neuronal subtypes in vitro requires exposure of these cells to morphogens and growth factors that recapitulate in vivo differentiation. In the case of MN differentiation, ES cells are stimulated with retinoic acid (RA) and sonic hedgehog (SHH) (41). ES cell-derived MN have all the molecular and functional characteristics of MN present in vivo (26), and when transplanted, can integrate (19) and cause a functional recovery in animal models of paralysis different from ALS (11).
In FALS models, some studies report a recovery in treated animals and a delay in the progression of the disease when human neural stem cells (hNSC) (42), human umbilical cord cells (16), or bone marrow stem cells (10) were used. In spite of the recovery observed with cell replacement strategies, it has become evident that MN degeneration in animal models of ALS is not a cell autonomous process (8). Several works have provided evidence that the environment created by nonneuronal cells could be detrimental to MN in animal models of FALS (5,13,29). ES cells comprise a hypothetically unlimited source in cell replacement therapies to generate specific cell types lost due to disease or injury. In addition, MN differentiation from ES cells is efficiently achieved by a well-established protocol. Therefore, the aim of this work was to differentiate mouse ES cells to MN that express green fluorescent protein (GFP), and to transplant them into the spinal cord of adult transgenic rats that express hSOD1G93A to evaluate their capacity to provide functional recovery.
Materials and Methods
Motor Neuron Differentiation Protocol
HBG3 mouse ES cells express GFP under the control of the hb9 promoter and are useful to identify neurons with a MN phenotype. HBG3 cells were differentiated as previously described (41). Briefly, HBG3 cells were expanded on mitotically inactive mouse embryonic fibroblasts cultured with knockout DMEM supplemented with nonessential amino acids, 2-mercaptoethanol, penicillin-streptomycin solution (Gibco, USA), 15% ES cell-tested fetal calf serum (Wisent, Canada), and 1000 U/ml of leukemia inhibitory factor (LIF; Chemicon, USA) to maintain their pluripotency. They were subsequently plated on gelatin-coated tissue culture plates and grown to 80% confluency whereupon they were tripsinized and seeded in suspension in bacterial plates to form embryoid bodies (EBs). Such EBs were cultured in ES cell medium for 2 days and subsequently treated with 2 μM RA (Sigma, USA), together with varying concentrations of recombinant human SHH (R&D Systems, USA), for 4 additional days. Finally, EBs were dissociated using a 0.2% papain/1 mM L-cysteine solution, and the resulting cell suspension was either reseeded on Matrigel-coated plates with DFNK differentiation medium consisting of DMEM:F12 and neurobasal medium (Gibco) supplemented with 2-mercaptoethanol, penicillin-streptomycin solution (Gibco), and knockout serum replacement (Gibco) to perform immunocytochemistry analyses, or used for transplantation studies.
Immunocytochemistry and Proportion of MN in Differentiated Cultures
To identify key markers of MN differentiation, immunocytochemistry analyses were performed on differentiated cultures as described previously (12,28,38). Cells were washed three times with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde for 20 min at room temperature. Cultures were washed three times with PBS and blocked by incubation with 10% normal goat serum/0.3% Triton X-100 in PBS for 45 min. The following primary antibodies were used at the indicated dilutions in 10% serum/PBS: rabbit anti-GFP 1:500 (Molecular Probes, USA), mouse anti-β-tubulin III antibody 1:1000 (Covance, USA), mouse anti-islet1 (Isl1) antibody 1:2 (Developmental Studies Hybridoma Bank, USA), and goat anti-choline acetyl transferase (ChAT) antibody 1:200 (Chemicon, USA). Corresponding secondary antibodies coupled to Alexa 488 or Alexa 568 (1:500; Molecular Probes, USA) were used. For ChAT detection, biotinylated anti-goat secondary antibody (1: 100), followed by incubation with Texas red-avidin (1: 200; Vector Laboratories, USA) was required. To label nuclei, 5 μM Hoechst 32258 dye was used. All experiments included cultures where primary antibodies were not added; unspecific staining was not observed in such negative controls. To estimate the proportion of MN induced by treatment of EBs with RA, alone or in combination with SHH, we quantified the percentage of GPF-positive cells from the total cell population in different experimental conditions: 1) 0.1% dimethyl sulfoxide (DMSO, RA vehicle); 2) 2 μM RA; 3) 2 μM RA + 20 ng/ml SHH; 4) 2 μM RA + 50 ng/ml SHH; 5) 2 μM RA + 100 ng/ml SHH.
Genotyping of hSOD1G93A Transgenic Rats
All animal procedures were approved by the local animal use and care committee and complied with Mexican and NIH guidelines. Transgenic hSOD1G93A animals were purchased from Taconic (USA). Nontransgenic littermates were used as controls. Male transgenic rats were mated with wild-type (WT) Sprague-Dawley female rats, and the resulting offspring were genotyped by the presence of hSOD1G93A through a genomic DNA PCR protocol recommended by the distributor. Rats were identified by subcutaneous implantation of a microchip (Allflex). Tail biopsies from 1-month-old rats were digested in proteinase K and then diluted 1:20 in sterile H2O, followed by heating at 95°C for 15 min. Two microliters were subjected to PCR amplification using primers specific for hSOD1 (forward: 5′-CGCGA CACAATCAAAGTGA-3′, reverse: 5′-CATGAGCCC TAATCCATCCATC-3′) to determine the genotype of the animals. In transgenic animals, we observed deterioration of motor abilities beginning with difficulty in posterior limb movement by 16 weeks of age and ultimately paralysis by week 19.
Transplantation of MN Derived From ES Cells
Transgenic and WT rats were transplanted at 10 weeks of age when hSODG93A animals were presymptomatic. One day prior to the surgical procedure, cyclosporin A (10 mg/kg of body weight daily) was injected IP, and this treatment continued daily until rats were euthanized. For transplantation, animals were deeply anesthetized with sevofluorane (5% for induction, 1% for maintenance) with the aid of a vaporizer. An incision was made above the lumbar region to expose the vertebrae. The vertebral column was secured to a stereotactic frame with the aid of clamps (Stoelting, USA) to perform laminectomy of L5. A small cut of the meninges was made to allow transplantation. Cell suspensions of differentiated ES cells were loaded on a microsyringe to unilaterally transplant 1 × 105 cells in 1 μl into the ventral gray matter (0.5 mm lateral to the midline and 1.5 mm ventral from the dorsal surface of the spinal cord). Sham rats were injected with 1 μl of DFNK medium. We performed the transplantation procedure in a cohort (n = 4) of transgenic hSOD1G93A rats that received cells previously labeled with 5 μM Hoechst 32258; these animals were sacrificed 1 week later to verify transplant location and to establish the presence of ChAT+ and GFP+ cells. In addition, the following groups were euthanized at 9 weeks postsurgery: WT sham (n = 4), WT grafted (n = 4), hSOD1G93A sham (n = 4), and hSOD1G93A grafted (n = 8). We performed histological analysis of these groups when transgenic hSOD1G93A were at the terminal stage of the disease.
Motor Function Test
In order to assess motor function the rotarod test was performed as described (9). This test consists of placing the rats on a rotating rod with an initial velocity of 10 rpm with an acceleration speed of 0.2 rpm/s until they fall off. Each week we recorded the time (in seconds) that the animals were able to sustain themselves on the rod, beginning at 14 weeks of age in sham and transplanted groups. This trial allowed us to establish the onset of motor deterioration and a possible therapeutic window for functional recovery.
Tissue Preparation and Immunohistochemistry
In order to analyze the spinal cord of WT and transgenic animals (sham and transplanted groups), they were euthanized by a lethal injection of pentobarbital (70 mg/ kg of body weight) at 19 weeks of age. Animals were first perfused with physiological saline to eliminate blood, and then with 4% paraformaldehyde in PBS for fixation. The spinal cord was dissected to preserve only the lumbar portion. Tissue was postfixed with paraformaldehyde and cryoprotected in 30% sucrose. Coronal sections of 30 μm were made close to the injection site. We analyzed the spinal cord of transplanted animals by staining with 0.1% cresyl violet solution as described previously (9) and by immunohistochemistry for ChAT and GFP to localize transplanted cells, either 1 week after grafting or at terminal stage when animals were paralyzed.
Statistical Analysis
All in vitro data are presented as mean ± SEM from at least three independent experiments. The number of animals used per transplantation group was 4 to 8. We performed ANOVA followed by Fisher post hoc test, and considered significant differences when p < 0.05.
Results
MN Differentiation
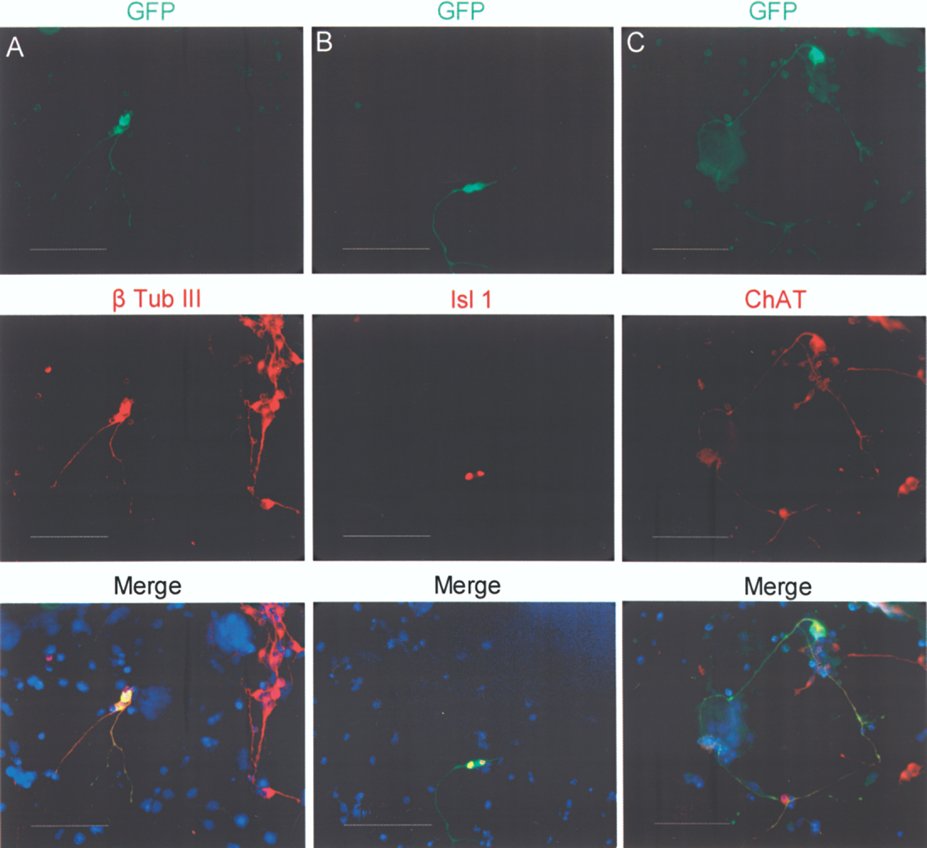
HBG3 ES cells were derived from transgenic mice that express GFP under the control of the MN-specific hb9 promoter. Previous reports demonstrated that treatment of EBs with RA and SHH induced MN differentiation of these ES cells (26,41). Treatment with 2 μM RA and 100 ng/ml SHH resulted in the appearance of GFP+ cells in the EBs (Fig. 1A, B), which upon dissociation and replating preserved GFP expression and neuronal morphology (Fig. 1C). In dissociated EBs, we quantified the percentage of GFP+ cells after incubation with DMSO (the vehicle for RA), 2 μM RA, or with RA plus increasing concentrations of SHH. In DMSO-treated cultures GFP+ cells represented less than 1% of total cells. This proportion increased with addition of RA only. In addition, we observed a significant dose-dependent increase in the proportion of GFP+ MN, reaching a peak at 16% of the total population with 2 μM RA and 100 ng/ml SHH (Fig. 1D). To further confirm the MN phenotype of GFP cells, we determined the presence of molecular markers characteristic of this neuronal type through immunocytochemical analyses. In cells dissociated from RA/SHH-treated EBs and plated on Matrigel-coated surface, we found that GFP+ cells coexpressed the neuronal marker β-tubulin III (Fig. 2A), and the MN transcription factor Isl1 (Fig. 2B) as well as ChAT (Fig. 2C), confirming that these cells were spinal MN.
Mouse ES cells differentiate to motor neurons with the addition of RA and SHH. Mouse HBG3 ES cells, wherein the hb9 promoter controls green fluorescent protein (GFP) expression, were used to form embryoid bodies (EBs), which upon treatment with 2 μM retinoic acid (RA)/100 ng/ml sonic hedgehog (SHH) contained GFP+ cells (A); phase contrast of the same field is shown in (B). This GFP expression (green) was preserved in cells with neuronal morphology after dissociation of EBs and replating. Nuclei were labeled with Hoechst 32258 and are shown in blue (C); images are representative of five independent experiments. Different conditions were assayed to establish the proportion of GFP+ cells after treatment with: 1) dimethyl sulfoxide (DMSO), vehicle for RA; 2) 2 μM RA; 3) 2 μM RA in combination with 20, 50, or 100 ng/ml of SHH. Significant increases in the proportion of GFP+ cells were found when EBs were treated with 2 μM RA/50 ng/ml SHH and with 2 μM RA/100 ng/ml SHH (D). Mean ± SEM from three independent experiments. *p < 0.05 relative to DMSO. Scale bar: 50 μm (applies to A–C). Motor neurons differentiated from ES cells express characteristic molecular markers. HBG3 ES cells stimulated with 2 μM RA and 100 ng/ml SHH differentiate to MN in vitro, as evidenced by several markers, including Hb9 (through the GFP reporter, A–C), which is coexpressed with β-tubulin III (β Tub III, A), islet1 (Isl1, B), and choline acetyl transferase (ChAT, C). Blue labeling in the merged images identifies nuclei stained with Hoechst 32258. Images are representative of five independent experiments. Scale bar: 100 μm.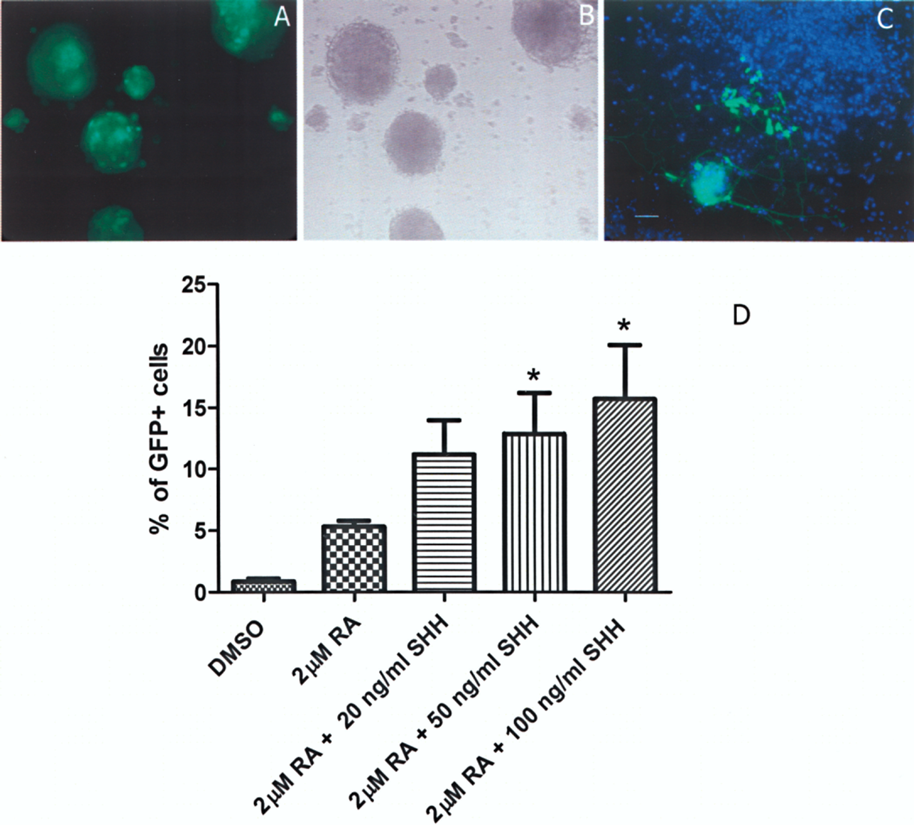
MN Transplantation
Cells obtained from EBs after 2 μM RA/100 ng/ml SHH treatment were used to transplant transgenic hSOD1G93A rats at 10 weeks of age, when no motor alterations were apparent. Animals received 1 × 105 differentiated cells in the ventral horn of the lumbar spinal cord (below L5 vertebra). In parallel, transgenic animals were injected with cell culture medium (sham group) in the same spinal region. In order to verify the injection site, cell suspensions labeled with Hoechst 32258 were introduced into transgenic hSOD1G93A recipients. Seven days later, four animals were sacrificed and the lumbar spinal cord recovered to obtain coronal sections. Labeled cells were located in the ventral portion of the spinal cord, close to the ontogenic site of MN (Fig. 3A, B). Transplanted cells were visualized by nuclear Hoechst staining. Some of these cells were GFP and ChAT double positive, indicating their foreign origin and MN identity respectively (Fig. 3C–F). Close to the graft, we also found ChAT+ Hoechst− GFP− endogenous MN (Fig. 3F).
Survival of transplanted MN derived from ES cells in the adult spinal cord of hSOD1G93A rats at 10 weeks of age analyzed 1 week postgrafting. Cells were incubated with Hoechst prior to transplantation and found 1 week later located in the ventral portion of the spinal cord, close to the ontogenic site of MN (A). In (B), a schematic representation of the lumbar spinal cord is shown with the area shown in (C–F) indicated by a red square. By confocal microscopy, we observed Hoechst-labeled cells (C) in these animals; some cells expressed ChAT (D) and GFP (E) as evidenced in the merge image (F). Images are representative of a cohort of four experimental subjects. Scale bar: 300 μm (A) and 100 μm (C–F).
Rotarod Test
We then examined the therapeutic capacity of ES cell-derived MN to prevent motor alterations in transgenic rats that resemble FALS. The rotarod test was employed to evaluate the motor performance of sham or transplanted WT animals. We found that both groups remained on the rotating rod for over 100 s at all analyzed ages regardless of having received ES cell-derived MN transplants or not (Fig. 4). In sharp contrast, sham-operated hSOD1G93A rats showed a clear decline in their capacity to remain running after 16 weeks of age and they were finally paralyzed by week 19. Paralysis generally commences in a hindlimb after 16 weeks of age, and this causes a decreased performance in the rotarod test. Interestingly, the transgenic transplanted group showed a significant recovery of motor function (Fig. 4) on weeks 16 and 17 maintaining on the rotarod 130 ± 8 and 130 ± 3 s, respectively, whereas sham animals remained for 91 ± 12 s (week 16) and 65 ± 15.5 s (week 17; p < 0.05 between sham and grafted groups for both time points). However this recovery was transient as MN transplanted animals eventually showed a decreased performance on the rotarod test, and finally became paralyzed, following a trend similar to sham group. At this stage animals were euthanized to perform histological analysis.
Transient motor recovery assessed by rotarod test after MN transplantation in transgenic hSOD1G93A rats. Surgeries were performed when rats were 10 weeks old for all groups. Starting at week 14, animals were placed on a rotating rod with constant acceleration to measure the time animals were able to sustain themselves by walking/running. Wild-type (WT) groups showed no decline in motor performance up to 19 weeks of age. Sham hSOD1G93A animals showed a decreased time on the rod commencing at week 16 and were completely paralyzed by week 19. Transplanted hSOD1G93A animals were indistinguishable from WT at weeks 16 and 17, and remained on the rod significantly longer than sham hSOD1G93A rats, but rapidly declined and were paralyzed at week 19 of age. Mean + SEM from four animals for WT sham, WT grafted, and hSOD1G93A sham and eight rats for hSOD1G93A grafted. *p < 0.05 relative to sham hSOD1G93A animals.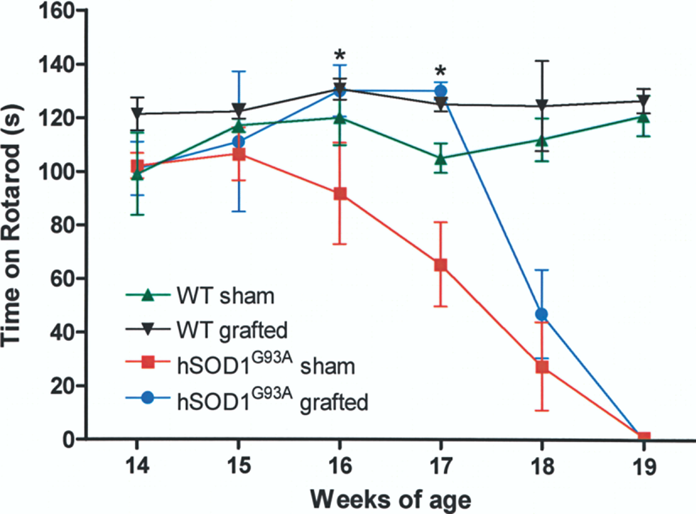
Histological Analysis of Spinal Cords
We analyzed the spinal cords of sham WT (n = 4) (Fig. 5A) and transgenic animals either with sham surgery (n = 4) (Fig. 5B) or grafted with MN (n = 8) (Fig. 5C) by cresyl violet staining at week 19 of age. MN were apparent by size and anatomical location in the ventral horn in sham WT rats, whereas the number of surviving MN in hSOD1G93A transgenic animals fell dramatically (remaining cells pointed with arrows in Fig. 5B and C). In grafted groups, we sought surviving MN in hSOD1G93A paralyzed animals and did not find GFP+ nor ChAT+ cells in the transplantation site (Fig. 5D). To rule out transplant degeneration or rejection in the spinal cord, we analyzed the presence of MN in WT animals. In contrast with findings in transgenic rats, we found ChAT/ GFP double positive MN in WT animals grafted at 10 weeks of age and sacrificed when they were 19 weeks old (Fig. 5E, F), demonstrating that these cells can survive for more than 2 months in a nontransgenic environment.
Transplanted MN survived 9 weeks in WT spinal cord, but not in transgenic hSOD1G93A environment. At 19 weeks of age, we performed cresyl violet staining in rats evaluated in rotarod and found surviving MN identified by their large somata in the spinal cord of sham WT rats (A). At the same age, paralyzed sham hSOD1G93A (B) and transplanted hSOD1G93A (C) animals showed a marked decrease in the number of MN with only a few of these cells surviving (arrows in B and C). Ventral side is at the top in these micrographs. In paralyzed hSOD1G93A grafted rats, we did not find GFP+ nor ChAT+ cells by confocal microscopy, indicating the loss of both transplanted and endogenous MN in the lumbar spinal cord (D); inset shows a phase contrast picture of this tissue, where the ventral portion is to the right. When we analyzed transplanted WT rats 9 weeks after grafting, we observed GFP+ (E) and ChAT+ (F) cells. In these animals, we found endogenous MN that were ChAT+/GFP− (indicated by arrows in E and F). Ventral is to the left in these last two images. Scale bar: 100 μm and 300 μm for inset in (D).
Discussion
The main goal of this work was to test whether ES cell-derived MN transplants prior to disease onset could ameliorate paralysis in a rat model of FALS. We present evidence that MN differentiated from mouse ES cells, transplanted into the spinal cord of adult hSOD1G93A transgenic recipients, cause a significant yet transient recovery in motor performance. These animals become paralyzed and neither endogenous (ChAT+ GFP−) nor transplanted MN (ChAT+ GFP+) were found at end-stage. In WT rats, transplanted MN survive for 9 weeks, and continue to express ChAT and GFP.
First we examined the capacity of cultured ES cells to differentiate into a MN phenotype with the addition of two morphogens that participate in their differentiation in vivo: RA and SHH. We observed that after stimulation of EBs with 2 μM RA and 100 ng/ml SHH, differentiated cells expressed markers indicative of MN commitment, such as β-tubulin III, Isl1, and ChAT. Furthermore, HBG3 cells used in this work express GFP under the transcriptional control of the hb9 promoter, which upon differentiation allow identification of MN by GFP expression. We found that close to 20% of cells were GFP+, a proportion similar to that previously reported (11,41). Miles et al. (26) demonstrated that MN derived from HBG3 ES cells can establish functional synaptic contacts with muscle cells in vitro. However, no reports of ES cell-derived MN transplantation into FALS rats have been published to date, despite the fact that several types of stem cells have been assayed on models of spinal cord injury (22).
Once we confirmed that the differentiated cells had the characteristics of MN, we explored their capacity to integrate into the adult spinal cord of hSOD1G93A transgenic rats. In order to later locate the transplanted population, we incubated the cells with Hoechst dye prior to transplant. Seven days later we found transplanted cells in the ventral horn of the spinal cord. These cells were positive for GFP, indicating their foreign origin, and for ChAT, confirming their MN identity. This suggests that transplanted MN could stop motor deterioration in transplanted transgenic animals by functionally restoring lost MN. Interestingly, our results indicate that in hSOD1G93A rats, transplanted cells caused a significant recovery as evaluated with the rotarod test during weeks 16 and 17 of age (6–7 weeks after transplantation), when compared to sham animals. This improvement was transient, as at later stages all transgenic animals showed a decrease in rotarod performance, and finally succumbed to paralysis. Several groups have transplanted stem cells in order to improve the motor deficits of transgenic rodents that express hSOD1G93A. Yan et al. (42) demonstrated that hNSC transplanted into the spinal cord of hSOD1G93A mice caused a functional recovery. They found that transplanted cells can differentiate to neurons and integrate with other endogenous neurons. Bone marrow transplantation also induces a recovery in hSOD1G93A mice (10). These results demonstrate that transplantation of stem cells into FALS models causes recovery and increase life span. On the other hand, similar to our findings, the effects published in these studies were effective only for a short period of time.
The transplantation procedure used here requires cutting meninges to inject the cell suspension, allowing the entrance of immune cells to the central nervous system. Xenografts are rejected by the host immune system through cellular responses (4). To prevent degeneration of transplanted cells, several immunosuppressive or immunomodulator molecules have been employed (4,42). Calcineurin inhibitors such as cyclosporine A prevent the action of effector T cells. Transplantation of stem cell progeny from human (40) or mouse origin (12,23,32) in rat brains have shown consistent and long-term graft survival when cyclosporine was used as monotherapy even transiently (40). The situation is different for intraspinal transplantation of transgenic rodents with human cells, where reports have shown variable (24,36) or no graft survival (42) in animals receiving only cyclosporine; combined immunosuppression or anti-CD4 antibodies promote human xenograft survival in hSOD1G93A mice (42). Other reports, however, show consistent mouse ES cell-derived xenograft implantation in the viral-damaged spinal cord (11,19), or the lesioned striatum of recipient WT rats immunosuppressed only with cyclosporine A (12,23,32). These findings, together with our results of consistent presence of grafts in WT rats 9 weeks posttransplantation, show that cyclosporine is useful in protecting grafted cells in the spinal cord of nontransgenic animals, and suggest that mutant hSOD1G93A rats are different to WT animals in host–graft interactions unrelated to immunological rejection.
Transplanted MN did not extend axons out of the spinal cord, and therefore did not reach muscles. We suggest that the observed preservation in motor function could be due to trophic support provided by the transplanted cells to the deteriorating endogenous MN. In line with this argument, it has been reported that human neural precursor cells engineered to secrete glial-derived neurotrophic factor (hNPCGDNF) and transplanted into the spinal cord of transgenic hSOD1G93A rats, caused increased cholinergic markers of MN (24). In addition, this same group observed that transplanted hNPCGDNF cells prevented degeneration of endogenous MN in the spinal cord, even at the final stage of disease. However, animals in this study showed also eventual paralysis possibly because the transplanted cells could not prevent degeneration of the neuromuscular junction (36). Also, it has been reported that hNPCGDNF cells were able to partially protect dopaminergic neurons when transplanted 1 week after a unilateral lesion was induced with the dopamine toxin MPTP in monkeys (14). Mouse NPC transfected with Neurotrophin 3 produced recovery in mice with dorsal spinal cord damage after transplantation (43). Bone marrow stromal cells, which include mesenchymal stem cells, have also been successfully used for the treatment of acute spinal cord injury with recovery probably mediated through the secretion of neurotrophins, axonal regeneration, or remyelination (30).
When we analyzed the spinal cords of paralyzed sham and grafted hSOD1G93A rats, we found a few MN evidenced by cresyl violet staining. At this stage, no MN were identified by immunohistochemistry for ChAT and GFP in grafted animals, suggesting that transplanted MN were also affected by the same hostile environment that cause degeneration of endogenous MN. In a previous report, it was shown that degenerating MN, stained by cresyl violet, were positive for ubiquitin while decreasing or losing ChAT immunoreactivity in FALS rats (36). Recently, it was recognized that other cell types can participate in MN death observed in transgenic rodents modeling FALS. Nagai et al. (29) demonstrated that conditioned media of astrocytes derived from hSOD1G93A transgenic mice caused MN death. There is also in vitro evidence suggesting that mutated hSOD1G93A microglia produce an increase of superoxide and peroxinitrite compared to the microglia of WT animals (5). In vivo studies also show that transplanted WT microglia can delay disease progression in transgenic hSOD1G93A rodents (5). Furthermore, in elegant chimeric studies, it was found that WT MN succumbed to cell death when surrounded by nonneuronal cells expressing mutant hSOD1G93A (8). In addition, a selective toxicity of MN has been demonstrated by in vitro studies where human ES cell-derived MN were incubated with glial cells isolated from animals that express hSOD1G93A (13). All these data show that the hSOD1G93A environment in the spinal cord is detrimental for endogenous and transplanted MN. This notion is further supported by the fact that we found GFP-expressing cells 2 months postgrafting in WT animals, indicating that transplanted MN can survive in nontransgenic animals. Other groups have reported similar findings with survival times above 6 months (11,19).
To achieve long-term recovery in transgenic hSOD1G93A rats, it is important to accomplish some basic conditions. First, cells must survive in the spinal cord of transgenic animals. One strategy to improve survival of these cells might be the cotransplantation of MN with mesenchymal stem cells. It is reported that these cells decreased astrogliosis and microglia activation that could be detrimental toward MN (39). Another possibility in the same direction is to optimize the in vivo survival of transplanted cells. For example, ES cells that express the antiapoptotic protein Bcl-XL were differentiated to dopaminergic neurons and these proved to be more resistant to toxic insults than dopaminergic neurons derived from WT ES cells (35). Also, viral delivery of vascular endothelial growth factor or insulin-like growth factor 1 in mutant hSOD1 transgenic animals delays paralysis onset and extends survival time (2,21). Secondly, another critical condition for achieving recovery is to create functional neuromuscular junctions. Desphande et al. (11) transplanted MN-committed ES cells into the spinal cord of animals paralyzed after MN depletion caused by Sindbis virus infection. Using a combination of MN graft, dibutyril cyclic AMP/rolipram (to inhibit myelinmediated repulsion of axon outgrowth) treatment, and transplantation of NPCGDNF into the sciatic nerve to promote axonal growth towards muscle, they observed that MN axons were extended outside the spinal cord innervating muscles, and accordingly, a long-lasting recovery in motor function 6 months after grafting. A third aspect to consider is the origin of cell deterioration. In FALS models, gene therapy has been shown to delay MN degeneration and to lengthen animal survival when interfering RNA designed to knock down hSOD1G93A in vivo was used; the fatal outcome, however, cannot be avoided (31). Therefore, it appears necessary to employ combinatorial approaches to prevent the degeneration of MN, or to carry out successful cell replacement therapy. All these strategies can, in the long term, be combined to design effective means of treating paralysis in ALS, or other forms of MN damage in vivo.
Footnotes
Acknowledgments
This work was supported by grants from PAPIIT, UNAM (IN224207), and Conacyt (14285). R.L. and P.K. received graduate fellowships from Conacyt. We thank Dr. Hynek Wichterle for donating HBG3 cells, Dr. Ricardo Tapia for allowing us to use the rotarod, and Dr. Anayansi Molina-Hernandez and personnel from IFC microscopy unit for technical assistance.