
Abstract
BACKGROUND:
Recent advances in medical care have increased life expectancy and improved the quality of life for people with Down syndrome (DS). These advances are the result of both pre-clinical and clinical research but much about DS is still poorly understood. In 2020, the NIH announced their plan to update their DS research plan and requested input from the scientific and advocacy community.
OBJECTIVE:
The National Down Syndrome Society (NDSS) and the LuMind IDSC Foundation worked together with scientific and medical experts to develop recommendations for the NIH research plan.
METHODS:
NDSS and LuMind IDSC assembled over 50 experts across multiple disciplines and organized them in eleven working groups focused on specific issues for people with DS.
RESULTS:
This review article summarizes the research gaps and recommendations that have the potential to improve the health and quality of life for people with DS within the next decade.
CONCLUSIONS:
This review highlights many of the scientific gaps that exist in DS research. Based on these gaps, a multidisciplinary group of DS experts has made recommendations to advance DS research. This paper may also aid policymakers and the DS community to build a comprehensive national DS research strategy.
Keywords
Introduction
Down syndrome (DS) is the most common chromosomal disorder in humans, affecting about one of every 675 births [1]. Underlying the diverse spectrum of phenotypes seen in people with DS is an extra copy of chromosome 21 (Chr21), or trisomy 21 (T21), which results in overexpression of many genes and changes in the proteome [2]. It is associated with intellectual disability, facial dysmorphism, short stature, and poor muscle tone or loose joints. There is also an increased risk of Alzheimer’s disease (AD), childhood leukemia, congenital heart disease, sleep dysfunction, metabolic disorders, autoimmune disorders such as thyroid disease, Type 1 diabetes, celiac disease, rheumatoid arthritis, and developmental disorders such as autism spectrum disorder (ASD) [3]. At the same time, however, individuals with DS have a decreased risk of developing atherosclerosis and certain adult cancers [4, 5]. With improved healthcare, people with DS are now living longer, with a life expectancy of > 55 years of age compared to just 25 years of age in the 1980’s. It is estimated that there are 210,000 people with DS in the United States and 40% are over the age of 30 years old [6].
Research to better understand DS across clinical, pathological, genetic, cellular, and molecular domains is essential to develop appropriate interventions to promote health and wellness across the lifespan. Such research has flourished in recent years, driven by technological innovations, emerging international collaborations, increased funding, and worldwide advocacy efforts. With the hope of leveraging the growing momentum to support DS research, two major DS organization, the National Down Syndrome Society (NDSS) and the LuMind IDSC Foundation crafted recommendations for a research strategy focused on the quality of life and care priorities of people with DS by 2030. The impetus for this effort was the US National Institutes of Health (NIH) RFI (Request for Information, Notice Number: NOT-HD-20-013) to update NIH research plans on Down syndrome in 2020. The NDSS and LuMind IDSC worked together with the scientific community to develop recommendations for the NIH. In addition, other DS organizations contributed, including the Jérôme Lejeune Foundation and the National Task Group on Intellectual Disabilities and Dementia Practices.
Methods
DS research is broad in scope and engages a wide spectrum of scientific and health care disciplines. To accomplish the goals of the project, eleven working groups made up of experts across multiple disciplines were established to focus on specific issues of special concern to people with DS. These topical groups addressed cognitive development; autism spectrum and behavioral disorders; impaired speech, language, hearing, and vision; heart and vascular disorders; sleep and respiratory problems; obesity, metabolic and musculoskeletal problems; cancer; immune system disorders; dental and oral health; and AD and aging. The eleventh working group focused on the importance of community engagement in all aspects of DS research to ensure that the strategy developed through this process is driven by and resonates with the DS community. Members of the community engagement working group came from a variety of organizations including LuMind IDSC, NDSS, local Down syndrome affiliates, and GiGi’s Playhouse. Important contributions were also made by caregivers and self-advocates to ensure that the DS community had input into the recommendations.
Workgroup members met via teleconference for over four months to discuss the state of the science in their respective topic areas, with plans to distill their findings into a research agenda for the next decade. In April 2020, a two-day meeting was held in a virtual format due to the COVID-19 pandemic. The virtual meeting stimulated discussion that enabled top researchers in the field to share information, look for areas of overlap across workgroup topics, and identify unaddressed needs and research gaps. This paper summarizes areas of medical need for people with DS that have been understudied, unfunded or underfunded historically by federal and state agencies and foundations. The article is intended to serve as a call to action to the entire biomedical research community, to act as a catalyst for advocacy in research on DS, and to give research on DS the attention and funding that people with DS deserve.
Cognitive development and independence
To understand the effects of T21 on the development and function of the brain, three main questions were identified as areas to prioritize: 1) What brain regions and cell types are affected and what is the relationship of these effects to cognitive phenotypes? 2) When do deficits arise; are they the consequence of neurodevelopmental changes or functional changes in neural cells; and are they preventable or potentially correctable? 3) What are the underlying mechanisms by which T21 causes cognitive deficits? By understanding the answers to these questions at cellular and molecular levels, it may be possible to identify therapeutic targets and at what point targeting them might prove useful to positively affect cognition.
Human studies provide information about how brain anatomy and structure change across the lifespan and are also critical for genomic analyses. Anatomical studies indicate that DS is associated with a smaller cortex and cerebellum, with reductions in the number of neurons but increases in numbers of glia [7–9]. Neurons have dendritic spine defects and altered synaptic plasticity while glial defects indicate altered myelination [10, 11].
Deficits have also been reported in the function of synapses, mitochondria, and endosomes; and cell stress pathways appear to be activated. Several human Chr21 (HSA21) genes have been implicated in development, including some that regulate neural development and developmental signaling pathways and others involved in multiple stress mechanisms. Many more HSA21 genes have not been well studied and gene interaction effects must also be considered. In addition, T21 affects the expression of genes throughout the genome and so consideration of molecular and cellular pathways, metabolic and immune defects, and environmental factors may also impact brain function in DS.
Translating these anatomical and neural findings has helped refine what aspects of cognition are targeted in studies of individuals with Down syndrome. Specific constructs of executive functioning, learning and memory, and working memory are being targeted with an emphasis on determining the best measures for assessing these constructs and change in these constructs [12–16]. Further attention to the measurement of cognitive constructs has focused on social cognition, emotion recognition, and evaluating cognitive batteries [17–28]. The downstream impact of these cognitive constructs on functional outcomes has primarily considered children with DS, understanding the impact on formal education, and adaptive daily living skills [29–35]. Given a pattern of findings linking cognitive skills to fine and gross motor control [36–41], preliminary small pilot trials to improve cognitive outcomes have more recently focused on the impact of physical activity interventions [42–44]. Both behavioral and pharmaceutical interventions are also being piloted to improve cognitive outcomes in individuals with DS [44–51]. Research on individuals with DS has frequently focused on between-group differences in comparison to other individuals with intellectual disability [52–57]. This focus has helped identify the unique cognitive phenotype common in individuals with DS. Group differences have also elucidated the impact of comorbid medical conditions on cognitive outcomes for individuals with DS, specifically cognitive outcomes for children with DS with or without comorbid congenital heart defects, sleep challenges including obstructive sleep apnea (OSA), and autism spectrum disorder [58–75].
Cognition may have impacts on other aspects of life such as independence. For individuals with DS, the degree of intellectual disability is one possible factor that may restrict independence. Studies show that speech ability and training, and access to a medical home can improve independence for individuals with DS [76, 77]. Measures of adaptive skills and surveys show that independence decreases with age in DS, which is likely multifactorial and presumably related to increasing risk for dementia [78, 79]. Drawing from studies of individuals with intellectual disability, speech independence, autonomy, and self-management predict independence, while low physical fitness and changes in ADLs may indicate decreasing independence [80–82]. Importantly, independence for individuals with intellectual disability can be promoted through interventions including video prompts, video self-modeling, staff training, and the use of technology and remote support services [81, 83–85].
While studies in humans are the gold standard, two types of research models are valuable to address cellular and molecular aspects of T21 in the brain: trisomic mouse models, and human T21 induced pluripotent stem cells (iPSCs) [86]. Human stem cell research enables investigations of T21 in the human genetic background at the molecular and cellular levels and enables mechanistic investigation. Current understanding of DS neurobiology is derived largely from mouse studies (e.g. Ts65Dn, Tc1) [87, 88]. Support for new mouse models that will minimize non-HSA21 genetic changes are needed [89]. In addition, it is becoming clear that aspects of gene regulation and brain development and function differ between mice and humans, so integrating information from these complementary models is essential to understand trisomy neurobiology and development [90–92].
Many drug candidates tested are reported to improve learning and memory in DS mouse models; however, the relevance of these findings to humans remains uncertain. Human cell and organoid models may be useful for testing drugs that target cell-based pathologies. Biomarkers –including functional measures such as functional magnetic resonance imaging (fMRI), electroencephalography (EEG), and functional near-infrared spectroscopy (fNIRS) - are clearly needed to advance drug development.
Cognitive development and independence research gaps
To gain a better understanding of cognitive deficits associated with DS, new preclinical models of cognition are needed to help establish the stage of cognitive development that could be amenable to improvement. Therefore, inducible models of trisomy silencing or specific gene silencing should be developed. In addition, mouse studies are needed that can establish a correlation between changes in synaptic plasticity with learning and memory deficits. Finally, new DS models should reflect human physiology and genetic heterogeneity (e.g. outbred backgrounds).
More support of human cognition research in DS is also needed. To establish the link between cognitive performance and underlying brain structure, neurocognitive tools such as fMRI, EEG, and transcranial magnetic stimulation (TMS) should be utilized. Reliable and valid clinical outcome measures across the lifespan should be developed, as well as studies on the natural development of these associated constructs, to enable longitudinal studies and to advance clinical trials [12, 15]. It is important to support research focused on the wide heterogeneity of cognitive function across the DS population, including the effects of medical conditions (e.g. Attention-Deficit Hyperactivity Disorder (ADHD), ASD, anxiety, Acute Myelocytic Leukemia (AML), Acute Lymphocytic Leukemia (ALL)) in order to identify those individuals who will benefit from a particular intervention and who would be appropriate for inclusion in a clinical trial. To better understand all the factors that impact cognitive skills and meaningful functional outcomes, it is important to establish strong collaboration across behavioral disciplines.
There is need to include assessments of independence when studying human cognition. To understand the factors that contribute most to independence, additional research on the natural history of independence in people with DS is needed. Specifically, consideration should be given to what aspects are most meaningful to an individual with DS and caregivers, how to measure independence reliably and validly, and how to modify those factors. Successful interventions used in individuals with ID can guide implementation and study in individuals with DS. Large, multi-site, longitudinal studies could evaluate human cognition and independence simultaneously to connect the lessons learned from basic science to clinical significance; with the ultimate goal of improving independence.
Behavior and autism spectrum disorder
The prevalence of coexisting psychiatric or serious maladaptive behaviors in children and adolescents with DS is high and the consequences on education, family functioning, and socialization substantial. It has been reported that coexisting neurobehavioral and psychiatric disorders in those with DS range from 18 to 38% [93]. In a Swedish population-based study of children and adolescents with DS, 42% were diagnosed with ASD and 34% with ADHD [94], with the severity of ASD but not ADHD positively associated with the level of intellectual disability [95]. Other studies have found similar rates of ASD and behavioral symptoms in children with DS [96].
Some adolescents and young adults with DS also present with progressive and sometimes rapid cognitive deterioration or regression, which may dramatically impair independence and autonomy [97, 98]. A minority of individuals with DS experience worsening autistic characteristics that progress to a dementia-like state in what has been called Down syndrome disintegrative disorder (DSDD) [99, 100]. The causes of regression and DSDD remain unclear, but may be associated with depression, hypothyroidism, autoimmune disorders, OSA, and/or subclinical epilepsy. One small study, which hypothesized an immune-related etiology, demonstrated significant improvements following treatment with immunotherapy [101].
Gaps in behavior and ASD research
Population-based normative data on maladaptive behaviors and psychiatric syndromes across the lifespan in DS are needed from studies with large and diverse cohorts. In addition, population-based studies are needed to evaluate the associations between ADHD, ASD, anxiety, depression, movement disorders, seizure disorders, and sleep disturbances. People with DS should be included in clinical trials of emerging therapies (e.g. novel biologics, anti-immune, anti-inflammatory, GABA/glutamate, and cell-based therapies) for autism and behavioral disorders. However, validated diagnostic and outcome measures need to be developed in order to assess behavioral disorders and autism for the DS population. In addition, neuroimaging and neurophysiology studies of behavioral disorders and ASD should be conducted in people with DS. Finally, there is a shortage of trained providers and researchers knowledgeable about DS. This has had a negative impact on advancing understanding of behavioral and psychiatric disorders in DS. Additional support for training fellowships in DS is needed.
Communication, vision, and hearing
A variety of structural and functional vision, hearing, and communication deficits are associated with DS, as well as other anomalies of the auditory/vestibular system. Variation in these structures and functions can be influenced by other aspects of the behavioral phenotype associated with DS. Taken together, these problems can interfere with the development and maintenance of communication skills critical to reading and oral language.
Communication disorders in DS
Difficulties with communication often occur in individuals with intellectual disability, but people with DS are 2.6 times more likely to have moderate communication difficulties and 1.9 times more likely to have severe communication difficulties than people with intellectual disability not associated with this syndrome [102]. It is important to understand the reasons why many individuals with DS have lifelong problems with oral and written communication. Individuals with DS often exhibit delays in language development, with greater delays in expressive than receptive language [103]. Difficulties with language development are demonstrated from very early in development and continue into adulthood, with delays observed in the earliest stages of prelinguistic communication. These delays continue into the adolescent and young adult years, including the use of complex language for participation and social interaction in everyday life [104]. Speech is the main form of communication in 97% of all people with DS and therefore is critical to social interactions of all kinds. Reduced speech intelligibility is common in children and adults with DS [105, 106], resulting from a combination of motor impairments, phonological delay or disorder, hearing loss, and craniofacial and laryngeal dysmorphology [107]. Speech intelligibility and language delays are major concerns for children with DS and their families. But many adults with DS also experience lifelong difficulty with the intelligibility of their speech [108]. Nonetheless, children with DS do demonstrate gains in language development and a pattern of relative strengths and weaknesses in this domain, including more substantial delays in expressive syntax than in expressive vocabulary, for example [109]. Recent studies have also shown that verb usage is particularly limited in people with DS, leading to less language complexity and poorer discourse skills [110]. However, there are developmental changes in this profile over time [103, 111]. For example, receptive and expressive vocabulary have been shown to improve during early adolescence but to decline in late adolescence and early adulthood, perhaps foreshadowing the later more dramatic declines associated with early onset Alzheimer’s disease. Given nuances in language for people with DS, treatment and intervention for language impairments in this population must consider both the language phenotype and overall behavioral phenotype [103, 112], leveraging language strengths to support treatment and intervention efforts.
Gaps in communication research
Research focused on the development of assessments and new interventions for improved communication for individuals with DS across the lifespan is needed. Validated, standardized measures of language may be used but may need modification for extensive testing in the DS population. The development of DS-specific language norms is needed, with the incorporation of other psychometric features, especially those relevant for use as outcome measures in treatment studies, including clinical trials. Studies that can distinguish the effects of motor speech disorders from effects caused by anatomical dysmorphology are needed to develop tools for clinical assessments and to facilitate the development of effective interventions across the lifespan. More and larger studies assessing the efficacy of high-intensity speech interventions in young children are needed. There is also a need to more fully understand the ways in which the social and linguistic environment supports or hinders language development in order to identify potential pathways and mechanisms for intervention. Lastly, there is a need for studies on language and communication in adulthood to understand the trajectory of improvement and possible age-related decline.
Vision disorders in DS
Ocular disorders are common in DS with reduced visual acuity observed even with refractive corrections. Specific impairments in vision include elevated refractive errors, strabismus, nystagmus, and corneal abnormalities all of which may contribute in part to the reduced visual acuity observed in DS [113]. These disorders are associated with abnormal visual developmental and possibly structural differences in the retina, cornea, and optic nerve [114–117]. Improved clinical strategies are needed to address visual deficits in this group, as most individuals do not achieve normal visual performance with standard refractions. Early intervention is likely to aid visual development for children with DS, but structural deficits in ocular tissues, such as those commonly observed in the cornea, may progressively worsen in later years, leading to additional losses in vision quality.
Gaps in vision research
A greater understanding is needed regarding the causes of reduced visual acuity, e.g., the contributions of retinal, corneal, and neural abnormalities. There is also a need to expand studies of the impact of optimized refractive correction on visual outcomes to younger children. Longitudinal studies with cohorts large enough to achieve statically meaningful results are needed to understand the progression of structural changes in the ocular system and correlations with visual outcomes across the lifespan.
Hearing loss and vestibular problems in DS
Hearing loss is common in children and adults with DS. The incidence of hearing loss in neonates and infants with DS is between 15% and 30% [118, 119]; between 25% and 85% in children and adolescents [120, 121]; and between 50% and 75% in adults [122, 123]. While most hearing loss in DS is associated with otitis media with effusion, mixed conductive-sensorineural loss is also common and is usually secondary to inner ear malformations [124], which may also cause symptoms of vestibular dysfunction such as dizziness, vertigo, and balance disorders. Anomalies in the auditory system in DS have been reported for the outer ear, middle ear, and inner ear [125–131]. Other factors that may contribute to vestibular dysfunction in DS include: hypotonicity, joint laxity, decreased deep tendon reflexes and delays in reaction timing, and equilibrium reactions. The vestibular system works with other senses such as vision and proprioception to maintain balance and motor coordination.
Gaps in hearing loss and vestibular function research
More research on the detection and treatment of hearing loss across the lifespan of individuals with DS is needed. Assessment and detection measures are needed that account for developmental and aging effects as well as structural differences that may be present. Research on strategies to increase the use of hearing aids among adults with DS who has hearing loss also is needed. There is evidence that DS-AD is sometimes misdiagnosed since the symptoms observed may actually be due to hearing impairment or other sensory deficits [132]. Hearing loss may also be a contributing factor given that it is more frequent among adults with DS and co-morbid dementia [133]. More research on the diagnosis of hearing impairment in adults with DS is needed.
Heart and vascular
Congenital heart disease (CHD) represents one of the cardinal features of DS, affecting more than 40 percent of infants with T21 [134]. Multiple subtypes are seen, including complete atrioventricular septal defects (AVSD), ventricular septal defect, atrial septal defect, partial atrioventricular septal defect, Tetralogy of Fallot, and patent ductus arteriosus; and the presence of multiple anomalies is common [135–139]. Early diagnosis improved surgical outcomes, and better perioperative care have resulted in significant increases in survival over the past two decades, contributing to the improved longevity for people with DS.
Trisomy 21 clearly significantly increases the risk for AVSD and other heart defects but is not sufficient to cause CHD. It was originally hypothesized that T21 unmasks a common susceptibility variant that explained the hugely increased in risk for AVSD observed in DS. Using a genome-wide association study approach, no evidence for this hypothesis was obtained [140–142]. Instead, current findings suggest an increased burden of rare variants as contributing to risk for AVSD in DS, some of which overlap with those found for non-syndromic CHD (nsCHD). Such variants (including single-nucleotide variants, microRNAs, and copy number variants) are not restricted to chromosome 21, but found throughout the genome [140, 143–145]. Thus, the modified hypothesis states that having an extra Chr21 predisposes to abnormal heart development, but additional rare variants or environmental triggers are required to exceed a susceptibilty threshold to lead to CHD. Support for this hypothesis and proof-of-principle has been provided by the Ts65Dn mouse model [146, 147]. Evidence for altered pathways include the ciliome, Notch signaling, VEGF-A and folate/homocysteine metabolism [144, 148].
In addition to CHD, individuals with DS often experience other cardiovascular problems including cardiac arrhythmia, pulmonary hypertension, and sleep apnea. A higher risk of cerebrovascular events including stroke and transient ischemic attack is also seen, particularly in women [149]. Immune dysfunction and thyroid dysfunction –both prominent in DS –may also affect cardiovascular function; and extra-cardiac comorbidities such as differences in vascular resistance and arterial stiffness may contribute to cardiovascular dysfunction. Alterations in heart rate variability and blood pressure in the low normal range are also relatively common in DS and may reflect autonomic dysfunction [150]. The consequences of lower blood pressure and heart rate in DS over the lifespan are poorly understood, as is the effect of exercise on autonomic function. The Ts65Dn mouse model also demonstrates reduced blood pressure and heart rate variability alterations from wild-type mice, suggesting this model could be used to explore mechanistic changes in cardiovascular function across the lifespan in DS [151]. Moyamoya disease may also occur in people with DS [152] and contributes to increased stroke risk over the lifespan, particularly with those who have blood pressure in a higher, but normal range [153].
Heart and vascular disease research gaps
The genetic factors and dysregulated pathways in DS-associated CHD are just beginning to be identified. The roles of nuclear and mitochondrial genes and their possible interactions with HSA21 genes have yet to be determined. Research integrating transcriptomic, metabolomic, proteomic and other -omic approaches should be applied to these knowledge gaps. In addition, epigenomic approaches could facilitate identification of environmental exposures associated with CHD. The integration of -omics data could help identify underlying molecular mechanisms, help define genotypes and phenotypes which could lead to prevention and novel treatments. From the clinical and epidemiological research perspective, larger sample cohorts with well-defined heart phenotypes (cardiac and extra-cardiac) are needed to define genotype / phenotype correlations in DS-associated CHD. Complementing these studies with model systems is essential to understand genotype functions and their interactions with the environment.
More clinical research is needed to better understand cardiovascular disease in DS. For example, there is a need to advance understanding of the surgical and postoperative care needs of children with DS and CHD. The early detection of Moyamoya disease is essential for better outcomes but this will require a better understanding of early risk factors and the development of novel biomarkers. The role of lifestyle factors such as sedentary lifestyle and exercise on cardiovascular function in individuals with DS is not well understood. There is early evidence that increasing exercise, even passive exercise, may also have positive effects on cognition and learning [44, 154]. One issue that needs to be addressed to advance clinical research in DS is the issue of control groups. Proper controls are essential for cardiovascular research in DS, however, there are many different types of controls utilized (heart rate controls, intellectual disability without DS controls, BMI controls, activity level controls), which complicates interpretation of data from these studies. The field should address this issue and build consensus around a single type of DS control.
Sleep and respiratory
Individuals with DS are prone to develop OSA due to a combination of anatomic and neuromotor factors, such as midfacial hypoplasia, macroglossia, tracheal and laryngeal abnormalities, and hypotonia [155, 156]. With aging, the prevalence of OSA increases but it remains frequently underdiagnosed. Untreated sleep disorders such as OSA may contribute to worse cognitive function and accelerate cognitive decline [45, 47]. Other respiratory problems including increased risk of infection are common in children and adults with DS due to a combination of the mentioned anatomical and functional abnormalities, immune dysfunction, and cardiac problems [157, 158]. For example, respiratory syncytial virus (RSV) infection is particularly prevalent and associated with high morbidity and mortality in infants and children with DS [159]. Dysphagia, also common in DS, increases the risk of aspiration and recurrent pneumonia [160].
Children with DS also have an increased risk for developing multifactorial pulmonary hypertension [161] associated with hypoxemia, OSA, pulmonary hypoplasia, increased pulmonary vascular resistance, and increased hemodynamic stress. The sub-group of children with DS and CHD are at particularly high risk of developing pulmonary hypertension, but it can also be a consequence of respiratory distress syndrome [162]. Lower airway anomalies such as hypoplasia of the alveoli and other more distal structures have also been reported [163].
Gaps in sleep and respiratory research
Despite previous sleep research, normative data on sleep patterns in DS are still lacking. In children and adults with DS, research is needed to evaluate the association of a characteristic DS sleep phenotypes, i.e. reduced sleep efficiency, decreased rapid-eye movement (REM) sleep, and increased slow-wave sleep (SWS, also called non-rapid-eye-movement 3 (NREM3 or N3) sleep), with learning and behavioral difficulties in the context of baseline neurocognitive impairment.
Neurocognitive studies to evaluate the impact of sleep disturbances in adults with DS and its relationship to cognition, behavior, and quality of life are needed. Indeed, it is important to prospectively assess the impact of sleep disturbances on cognitive impairment and progression to dementia in adults with DS. Objective sleep evaluation methods and a DS-specific cognitive battery are needed to properly evaluate baseline relationships and intervention outcomes. More research is required on circadian rhythm disruptions in people with DS. Finally, the design and validation of new sleep questionnaires and simple at-home devices to screen for sleep disorders are needed.
Pulmonary hypertension is a significant cause of morbidity in children and infants with DS. More research is needed to distinguish the clinical and molecular pulmonary hypertension phenotypes in DS compared to children without DS. In addition, non-invasive airway evaluation guidelines for children with DS that incorporate multidisciplinary aerodigestive programs are needed.
Musculoskeletal, metabolic factors, and obesity
Past research findings indicate that individuals with DS across the lifespan are at high risk for musculoskeletal issues and metabolic disorders such as obesity and Type 2 diabetes [164, 165]. The genetic and molecular mechanisms that underlie musculoskeletal and metabolic dysfunction in DS remain unclear. T21 has been associated with altered insulin secretion, impaired hepatic glucose metabolism, and altered insulin sensitivity in muscle. Autonomic dysfunction with altered peripheral blood flow to muscle may affect blood distribution to the muscle and build-up of toxins, which may lower the pain threshold and tolerance for physical activity. Research in both humans with DS and DS mouse models has identified changes in metabolism related to obesity, inflammation, mitochondrial function, immunity, insulin resistance, and glucose tolerance [166].
Individuals with DS across the lifespan are at higher risk to be overweight and obese compared to the general population [167–170]. The possible determinants of increased weight and obesity in individuals with DS include low resting metabolic rate, low physical activity levels, and unhealthy dietary behaviors [168, 171–173]. Obesity appears to have adverse health outcomes among individuals with DS such as dyslipidemia, hyperinsulinemia, OSA, and gait problems [168, 174–178].
Musculoskeletal problems associated with DS include muscle hypotonia and joint laxity, both of which can cause ambulation difficulties and other functional impairments [164]. Moreover, these problems may contribute to reduced physical activity, increased levels of obesity [168], and an elevated risk of many disorders including heart disease, Type 2 diabetes, cancer, and osteoporosis [179]. Individuals with DS across the lifespan have very low physical fitness and physical activity levels [180–184]; these attributes may be either causes or outcomes of musculoskeletal and metabolic issues or bidirectional relationships may be at play. Certainly, improving the physical activity and physical fitness levels of individuals with DS may positively impact their health and functional profiles.
There are ongoing international efforts to better understand metabolic dysregulation in DS. The Gene Overdosage and comorbidities during the early lifetime in Down syndrome (GO-DS21) project recently launched in the European Union to explore metabolism and metabolomics in DS.
Gaps in musculoskeletal and metabolic research
Longitudinal studies on muscle development and weight changes across the lifespan in DS are needed to explore differences between people with DS and the general population. These studies should evaluate the development of obesity across the lifespan, delineate its causes and clinical impact, and test interventions for reducing obesity in individuals with DS. Hypotonia is very common in DS but it is not well understood. Research on the genetic and biochemical basis of hypotonia including the role of mitochondrial alterations is needed to clarify the etiology of the condition. In addition, the effect of impaired autonomic function on cardiovascular fitness is not well understood and needs more research. There have been a few small physical activity intervention trials in DS [185–187]. More and larger trials are needed to test the effects of physical activity as an intervention on a range of health outcomes and the modifications that may be needed to implement such interventions in the DS population.
Cancer
Although there is no difference in the overall incidence of cancer among the DS population compared to the non-DS population, DS is associated with a decreased incidence of solid tumors and an increased incidence of hematological malignancies, specifically acute leukemias [5, 188].
Compared to the non-DS population, individuals with DS are 500 times more likely to be diagnosed with acute myeloid leukemia (ML-DS) and 20 times more likely to be diagnosed with acute lymphoblastic leukemia (DS-ALL). Survival outcomes for ML-DS are superior to the general population, with event-free survival rates ranging from 80–100%. Clinical trials are focused on optimizing chemotherapy to reduce toxicity while maintaining survival outcomes [189, 190]. Outcomes for patients with DS-ALL are inferior to the general population and patients with DS treated for ALL have excess treatment toxicity and treatment morbidity. Novel approaches to treat DS-ALL are needed to improve survival and decrease treatment morbidity [191, 192]. Factors that contribute to variability in treatment response and outcomes are not fully understood but may include differences in the genetics and biology of DS-leukemia as well as those related to the DS-phenotype (i.e., gene dose effect from T21).
The risk for DS-leukemia (ML-DS and DS-ALL) is greatest during early childhood (ages 1–4), a critical neurodevelopmental period. Treatment for DS-leukemia (DS-ALL in particular) involves central nervous system (CNS)-directed chemotherapy. Treatment for DS-ALL is 2.5 –3 years in duration with extended periods of immunocompromise resulting in missed opportunities for social interaction, early intervention and education services that support developmental gains throughout childhood. It has been well-established that survivors of childhood ALL without DS have a higher risk of neurocognitive deficits [193, 194]. Given the preexisting cognitive vulnerability in DS, the cumulative impact of CNS-directed treatment and missed community participation may add to neurocognitive deficits. Compared to survivors of leukemia without DS, survivors of DS-leukemia may be at increased risk for treatment late effects and poorer quality of life [195–197]. Improved understanding of the impact of leukemia and its therapy on neurodevelopmental, health, and quality of life outcomes has the potential to inform modifications to treatment, approaches to supportive care during therapy, and interventions to ameliorate side effects.
Gaps in cancer
Improved characterization of clinical, biological, and genetic phenotype in DS-leukemia is needed to identify therapeutic targets and further refine treatment. A better understanding of DS-leukemia genomics may also have implications for surveillance and diagnosis. Evidence-based approaches to supportive care, improved assessment and management of side effects and toxicities in DS leukemia needs to be developed. Longitudinal studies of neurodevelopmental, health, and quality of life outcomes in cancer survivors beginning during therapy, in order to inform supportive care and interventions to ameliorate problems are also needed.
Immune system disorders
The architecture of the immune system is significantly altered in people with DS, with common findings of leukopenia, lower B cell frequencies, and pro-inflammatory shifts including increased proportion of memory T cells, pro-inflammatory T cell subsets, and pro-inflammatory cytokine-producing cells [198, 199]. Consistent with this pro-inflammatory milieu, people with DS exhibit a highly increased risk of developing autoimmune diseases including thyroid disease, Type 1 diabetes, celiac disease, rheumatoid arthritis, systemic lupus erythematosus, and atopy. Paradoxically despite this augmented immune response, individuals with DS are more susceptible to infectious disease, which account for 50% of deaths in individuals with DS [3]. This includes increased mortality secondary to RSV and now to COVID-19 infections. [198–204]. Additionally, these diseases may manifest at earlier ages and greater severity in people with DS.
Gaps in immune system disorders research
Critical gaps exist in our understanding of the specific facets of immune dysregulation and the underlying mechanistic pathways, which drive predisposition to autoimmunity and poor outcomes with infection. Research is needed to clarify which existing therapeutics are most effective for people with DS and to prioritize targets for novel therapeutics. Although candidate genes on Chr21, including IFNAR1/2 and DYRK1A, have been identified, the role of most HSA21 genes is unclear. Additionally, studies showing that T21 can impact transcription on other chromosomes suggests that the scope of this research will likely extend beyond Chr21. Because the risk of autoimmunity increases with age, longitudinal studies are also needed with genomic confirmation of full or partial trisomy of genes associated with immunity. Sampling prior to developing autoimmunity is necessary to allow us to better understand who is at higher risk, and how that risk might be mitigated. Genotyping for known autoimmunity-associated polymorphisms will help us better understand how T21 modulates single nucleotide polymorphisms (SNP)-associated risk. Similar SNPs on T21 that are normally rendered silent might exert their effect due to gene dosage, but also perhaps due to overall perturbed clinical state. Linked clinical metadata, particularly the severity of autoimmunity and response to specific therapeutics, will enhance translational significance of this work. To better understand the role of thymic dysfunction in autoimmunity, studies should assess the consequences of thymectomy (e.g. on the differentiation of T cells into subsets and the production of self-reactive T cells) in children with CHD who undergo this procedure to provide surgical access as part of their cardiac surgery. As the immune system penetrates all organ systems, it is important to better understand how the immune system connects to dermatological, neurological, and gastrointestinal systems and how it may influence the course of disease in cancer and AD alike. Finally, the emergence of the COVID-19 pandemic makes research in the DS population a priority including for the development of potential therapies.
Dental & oral health
Issues with dental and oral health are very common in DS and often directly impact the quality of life. There are many differences between individuals with DS and the general population [205]. Differences include delayed eruption in babies and children, differences in eruption sequences for primary teeth, microdontia, and hypodontia [206, 207]. People with DS may have large tongues (macroglossia), or they may have an average size tongue but a small upper jaw that makes their tongue too large for their mouth (relative macroglossia) causing difficulties with speech and breathing and contributing to OSA. Small jaws and microdontia can cause tooth crowding and problems with spacing where the teeth of the upper and lower jaws do not touch affecting the bite. Orthodontics may be able to improve some of these issues, but this may present a particular challenge for children with DS. Finally, there are reports that people with DS have a lower risk for cavities; however, much of that research was done when people with DS lived in institutions and had very restricted diets. As such, more research is needed on the caries prevalence in people with DS [208, 209].
People with DS may have an aggressive form of periodontitis characterized by rapid progression, significant bacterial and inflammatory burden, and an onset as early as 6 years of age, which could contribute to other systemic diseases as well [210–213]. However, studies have shown that periodontal therapies may be effective [214]. It has been observed that in elderly cognitively normal people, measures of periodontal destruction and periodontal dysbiosis is associated with brain amyloidosis [215]. Given the high prevalence of both AD and periodontal disease in DS, more research is needed to determine if periodontitis contributes to AD pathology [216].
Dental and oral health has a broad impact on overall health and quality of life. Thus, more research is needed to understand the impact of dental and oral health in DS and the association with development, sleep disorders, the immune system, and other common co-occurring conditions. This research could lead to greater insights in the role of oral health on the overall health of people with DS and lead to better treatment options.
Dental and oral health research gaps
The development and validation of cellular and animal models are needed to better understand the characteristics of dental and oral health in individuals with DS. More research is needed to better characterize the differences between DS and the general population in dental development and oral health to help inform medical guidelines. Furthermore, the impact of periodontal disease on the overall health of individuals with DS is not well understood. DS mouse models should be employed to study the possible association between periodontal disease and AD related pathology such as Aβ plaques, neurofibrillary tangles, and neurodegeneration. Longitudinal and interventional studies should evaluate the role of periodontal inflammation and bacterial dysbiosis on AD progression [217].
Alzheimer’s disease and aging
With improved care and extended longevity for people with DS, there has been an increase in age-related disorders, especially AD, which typically occurs at an earlier age than that observed in the general population [218] (Fig. 1). Indeed, compared to people without triplication of Chr21, individuals with DS are at a markedly increased risk of developing AD. By age 40 years, β-amyloid plaque pathology and neurofibrillary tangles are present in almost all people with full trisomy [219]. It is estimated that the lifetime risk of AD is > 90% and that AD is the leading cause of death for adults with DS [220, 221]. However, few adults with DS associated AD (DS-AD) or with other age-related disorders receive appropriate care for a number of reasons. These include a shortfall of accepted and validated standards for care, lack of practitioners trained to provide support and evaluate people with DS-AD, inadequate data on the natural history of persons with DS-AD, lack of DS-specific diagnostic and treatment approaches, lack of evidence of efficacy of existing treatments, and inadequate resources for caregivers.
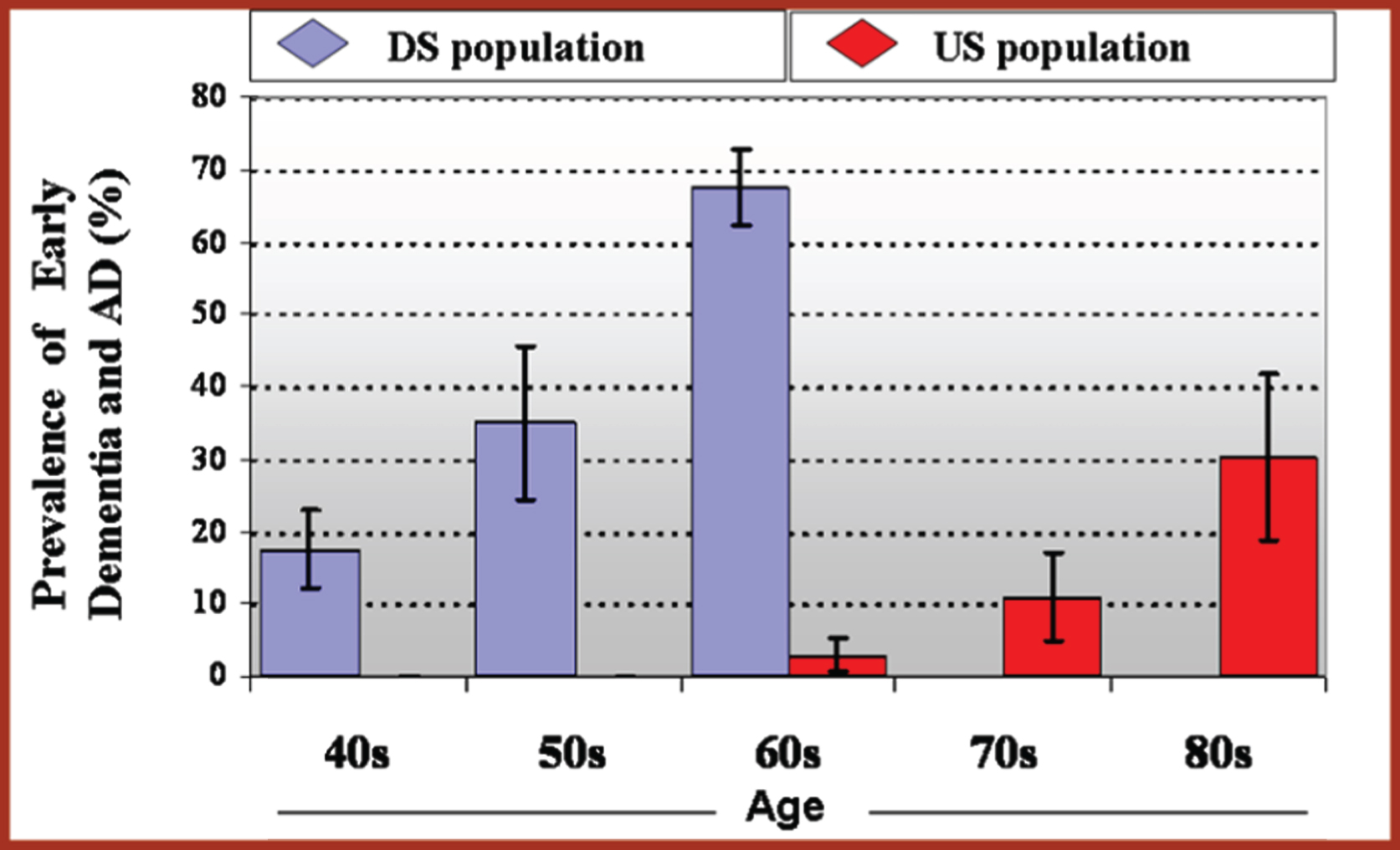
Prevalence of Early Dementia and AD by Age [218].
The reasons for the increased incidence of DS-AD are multifactorial but a primary driver is likely related to genes on HSA21. Among the genes expressed on HSA21 is the gene for amyloid precursor protein (APP). Overexpression of this protein due to trisomy leads to excess production of the toxic, AD associated protein, β-amyloid. β-amyloid accumulates within senile plaques in the brain but also can affect the blood vessels of the brain (cerebral amyloid angiopathy or CAA) [222]. Although most people with DS are rarely affected by atherosclerosis and arteriosclerosis, which are two risk factors for cerebrovascular pathology, there is significantly more CAA in DS than in people without DS or when compared to older people with AD [223, 224]. Neuroinflammation may also show unique signatures in the brains of people with DS, with features both common to and unique from sporadic AD [225]. Thus, there are multiple pathological events that occur either serially or in tandem to accelerate the development of AD in people with DS.
Genetic, cell biology, cellular/animal models, and natural history studies have provided insight into the aging process and the increased risk of dementia in individuals with DS. It is well known that the brain undergoes accelerated aging changes in DS, but biological mechanisms for this have not been fully explored. These studies suggest that increased expression of HSA21 genes induces many cellular changes, including dysfunction of endosomal and lysosomal systems, composition of neurotrophic signaling, increase in amyloid aggregation and cognitive defects, and alterations of microglia and astrocyte function [219, 226–232]. Genome-wide association studies (GWAS) have identified 25 candidate DS-AD risk alleles. In small DS participant datasets, longitudinal telomere length changes correlate with cognitive decline and accelerated epigenetic aging in DS is under investigation. Investigators probing the gut microbiome in people with DS have identified bacterial species that correlate with Aberrant Behavior Checklist (ABC) scores [233]. In addition, novel biomarker methods including neuron-derived exosomes and more sensitive plasma-based assays allow early diagnostics and a potential to better evaluate efficacy of interventions [234, 235].
Clinical phenotyping, biomarker development and validation in DS, and neuropathological diagnostic research have also progressed in recent years, enabling several clinical trials of potential AD therapeutics in individuals with DS. In addition, animal and cellular models in development have enabled exploration of therapeutic approaches, with early evidence that immune approaches may be feasible [236]. Many collaborations and partnerships have been established in recent years to accelerate research on DS-AD, including: Horizon 21, a multisite study in Europe recruiting a trial-ready cohort [237] The Alzheimer’s Biomarker Consortium-Down Syndrome (ABC-DS) –defining conversion to dementia based on biomarkers [238] Longitudinal Investigation For the Enhancement of Down Syndrome Research (LIFE-DSR) –an observational cohort study [239] Alzheimer’s Clinical Trials Consortium - Down Syndrome (ACTC-DS) is preparing to recruit a trial-ready cohort [240] 3-Star study of the candidate vaccine, ACI-24 sponsored by AC Immune is nearing completion; AC Immune will also soon launch a Phase 2 trial of β-amyloid vaccine in adults with DS[241]
More research support is needed to better understand the genetics and epigenetics of DS-AD. The underlying mechanisms that lead to high APP expression in DS-AD pathogenesis is not well understood. For example, genetic and epigenetic regulation of APP expression remains largely unexplored in people with DS. Epigenetic aging studies have been virtually unexplored in DS and could lead to novel genetic pathways that are amenable to intervention. Another yet unexplored area is the influence of specific microRNAs in the AD pathology. Chr. 21 contains several microRNAs, of which some have been shown to affect AD pathology. There is a need to perform sampling at multiple time points to evaluate trajectories across the lifespan. To explore the role of additional genetic AD risk factors, it will also be valuable to compare GWAS hits in DS-AD to those from late onset AD (LOAD) including their association with age of onset, cognitive decline, and variation in risk by sex or ethnicity. Other risk factors also need research attention such as sex, race, ethnicity, the role of inflammation, the immune system, and potential links to cerebrovascular pathology in DS-AD across the lifespan. There is a need to further refine the amyloid/tau/neurodegeneration framework in DS for comparison to LOAD and dominantly inherited AD and to identify where and how neuroinflammation and cerebrovascular pathology contribute. The efficacy of treatments and care models in DS-AD are knowledge gaps. The use of approved AD drugs in people with DS needs study to determine if these widely used drugs are safe and/or appropriate for DS-AD. It is unknown if the therapeutic window of approved AD drugs is different for DS-AD as compared with LOAD. In addition, new care models need to be developed that enhance the quality of life and longevity in individuals with DS-AD.
Community engagement
Community engagement is a continuum depending upon the level of collaboration between researchers and the community yet is essential since members of the DS community can give meaningful insight into issues that are important to them across the lifespan. Community members should thus be included in all aspects of research studies, starting with the design of the study, the determination of inclusion and exclusion criteria, the selection of outcome measures, the recruitment of participants, and the development of informed consent procedures. As a research study progresses, members of the DS community should continue to be included in discussions of the progress of the study, interim results, and communication protocols with research participants.
Community engagement research focuses on ensuring that the inclusion of community members in studies achieves the goals of improving their understanding of the purpose of research, optimizes recruitment for clinical studies, improves the reliability and validity of measurement tools, and improves the translation of research into practice. The Community Engagement working group recommended the need to communicate the message better so that people with DS understand the reason for the research. They also called for the development of strategies to increase participation of people with DS in clinical trials and to engage individuals with DS in the design of such trials.
Community engagement gaps
A recent survey of 256 individuals with DS found that 86% want new treatments and interventions, but 64% never participated in research studies. In addition, for those that have participated in research studies, direct interviews with individuals with DS and their caregivers found a lack of meaningful engagement with them at each step of the research process. Increased efforts are needed to better understand the factors that lead to individuals with DS being significantly under-represented and oftentimes excluded from research studies, including potential discrimination, rigid inclusion and exclusion criteria, low prioritization and limited community outreach and awareness. More infrastructure is needed for outreach and information sharing and dissemination. In particular, new outreach approaches are needed for the older adult community who tend to live outside the family unit in independent living, groups homes and long-term care facilities.
Overall Priority Recommendations for DS Research
Overall Priority Recommendations for DS Research
Each workgroup identified research needs for their specific focus area and many of these research gaps have been highlighted above. However, the workgroups also identified areas that represent clear DS research needs that cross multiple disciplines. The table below summarizes eight areas of overall research need that, if funded, would significantly improve the understanding of DS and lead to improvements in treatments and quality of life. The eight overall topic areas are clinical and genetic phenotyping, cellular and animal models of DS, longitudinal studies, randomized controlled trials in the DS population, centralized biorepository, open access centralized DS data, research training for clinicians and scientists, and research inclusion. The workgroups also developed a comprehensive list of recommendations in response to the DS RFI. The full list of recommendations can be found at: https://www.nih.gov/sites/default/files/research-training/initiatives/include/down-syndrome-RFI-responses-combined.pdf.
Conclusion
While it is true that the life expectancy for people with DS has increased significantly in recent decades thanks to advances in medical care, this review clearly demonstrates that many scientific gaps remain that prevent further advances in the discovery of new treatments and improvements in quality of life. These research recommendations were developed specifically in response to the NIH RFI and it is the hope of the coauthors that the NIH will not only include the recommendations submitted and published here in their Research Plan on Down Syndrome, but also provide sufficient funding for implementation. The coauthors also recognize that, for these recommendations to achieve their desired result, a coordinated approach with other funders of research, including other U.S. government agencies, and non-government organizations is needed. In addition, it is hoped that this paper will serve as a call to action for policymakers and the DS community around a focused and comprehensive national Down syndrome research strategy.
Dedication
This paper is dedicated to the memory of Dr. Angelika Amon who was a leader in Down syndrome research and who made significant contributions to this review.