
Abstract
BACKGROUND:
Targeted therapy using anti-TNF (tumor necrosis factor) is the first option for patients with rheumatoid arthritis (RA). Anti-TNF therapy, however, does not lead to meaningful clinical improvement in many RA patients. To predict which patients will not benefit from anti-TNF therapy, clinical tests should be performed prior to treatment beginning.
OBJECTIVE:
Although various efforts have been made to identify biomarkers and pathways that may be helpful to predict the response to anti-TNF treatment, gaps remain in clinical use due to the low predictive power of the selected biomarkers.
METHODS:
In this paper, we used a network-based computational method to identify the select the predictive biomarkers to guide the treatment of RA patients.
RESULTS:
We select 69 genes from peripheral blood expression data from 46 subjects using a sparse network-based method. The result shows that the selected 69 genes might influence biological processes and molecular functions related to the treatment.
CONCLUSIONS:
Our approach advances the predictive power of anti-TNF therapy response and provides new genetic markers and pathways that may influence the treatment.
Introduction
Rheumatoid arthritis (RA) is a complex autoimmune disease for which there is no cure. However, to relieve symptoms and prevent the disease from progressing, a variety of powerful treatments are available. TIn order to prevent permanent loss of function associated with structural damage to the joint, early therapeutic intervention is recommended [1]. For 90% of biologically untreated patients with RA, anti-TNF therapy provides the first effective treatment option if conventional synthetic disease-modifying antirheumatic drugs such as methotrexate do not work [2]. However, of these RA patients, *70% do not gain meaningful clinical change with anti-TNF treatment [3]. To predict which patients will not benefit from anti-TNF therapy, clinical tests should be performed prior to treatment beginning.
As genomic technologies have advanced, we have better understood inflammatory diseases and developed new treatments. Through the transcriptome, we can view specific genes over-expressed or under-expressed in diseases as a way to gain insight into a cellular response. Although various efforts have been made for identifying biomarkers and pathways [3], the specific response to anti-TNF therapy still remains unraveled. The statistical framework in most of these studies is based on a single set of data and does not take into account the knowledge in protein-protein interactions, biological regulatory networks and signaling pathways. In such a framework, the lack of biological information leads to the stability of prediction factors and reduces the predictive ability of the model [4]. In order to introduce modern precision medicine to autoimmune diseases, an advanced computational method combining genetic data with biological processes is needed.
There are many types of biological network information, such as functional interaction networks [5], protein-protein interactions (PPI) [6], correlations between genes [7, 8], KEGG pathways [9]. There are several studies that use biological knowledge, including those by Li and Li [10], Huang et al. [11], Wang et al. [12] and Chen et al. [13]. They described genomic knowledge as a graph that encoded genetic relationships (edges) among genes (nodes). Following that, they implemented linear and classification models with penalties based on Laplace matrices. Models that exploit biological information a priori are known as network-based approaches.
The hypothesis that complex diseases such as RA arise and develop due to interactions between several interrelated pathogenic genes, is supported by a growing body of evidence, indicating that the evaluation of the influence of any single variant is complicated [14]. This study hypothesizes that combining biological interaction information with gene expression data would help identify more robust biomarkers to predict the clinical response to anti-TNF treatment. Therefore, we tried to select the predictive biomarkers by using a network-based computational method to guide the therapy of RA patients. Our results have provided new candidate genes and pathways that may be predicting the response the anti-TNF therapy.
Method
In order to integrate the analysis of gene expression data with biological networks, we propose using the Laplace constraint method [10]. Let a network
where
The first term in Eq. (1) represents the loss function, secondly, network-based penalty provides a chance to capture interactive biological knowledge. Parameter
Equation (1) struggles in high-dimensional applications where the number of genes, is larger than the sample size [15, 16, 17, 18]. To solve the problem of large
where
Its higher estimation accuracy and Oracle property make it more advantageous than the lasso method. Therefore, we use the SCAD method to penalize the network-based methods as proposed in Eq. (5). Finally, the model we adopted in this article is defined as:
Where
and
To solve Eq. (4), we use the following coordinate descent method. More detailed information can be found in Eq. (5).
Step 1: Update
Step 2: Let
Data description
To identify the key clinical predictive biomarkers for RA, 46 samples with RA, including 24 response to anti-TNF therapy and 22 no response to anti-TNF therapy, were included in the study. Expression data from peripheral blood from these subjects were collected [20].
We mapped the dataset to an official gene symbol, and we calculated average expression levels for multiple probe sets mapped to the same gene. BioGrid provides the biological interaction network
Construct model and select biomarkers
Tenfold cross-validation on multiple dimensions was used to find the optimal regularization parameters of the model. A classifier model was constructed with the estimated tuning parameters and all the training data with 69 genes (Table 1) and perfect classification performances (Fig. 1). Among all the cutoff points, the one with the highest sum of sensitivity and specificity was chosen.
The selected 69 genes from chronic obstructive pulmonary disease gene expression data
The selected 69 genes from chronic obstructive pulmonary disease gene expression data
Training Performance. A1: ROC curve analysis; A2: test scores to be a case of all samples from the dataset were ranked. No response anti-TNF therapy cases are colored in green and response cases in red. 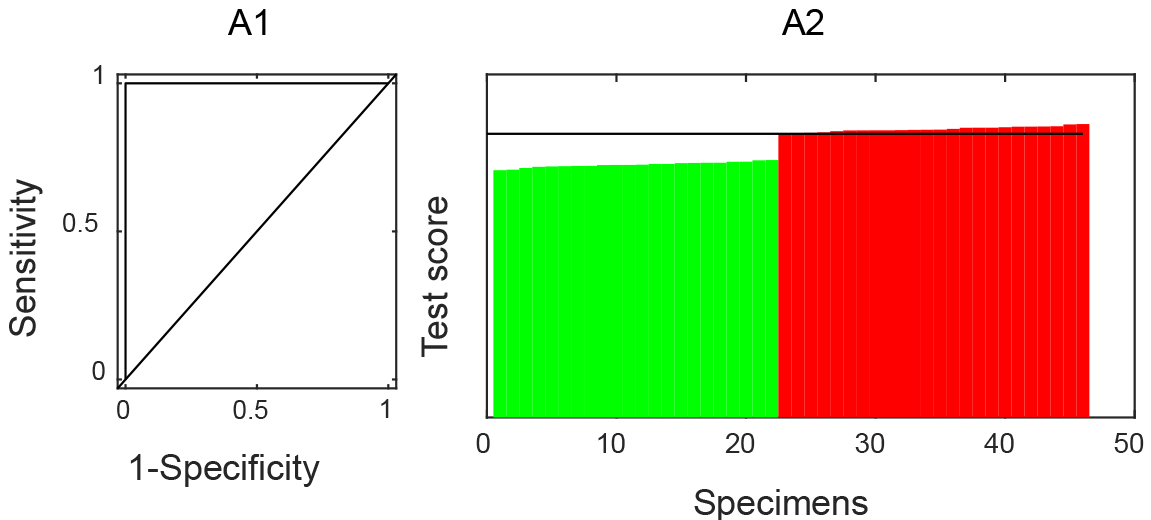
Among the 69 genes, there are some interesting findings. For example, Rui et al. [21] examined the contribution of CASP5 gene polymorphisms to RA risk in a Chinese population. They confirmed that CASP5 was related to the development of inflammation, which is the main feature of RA. Thus, through its role in mediating inflammation, CASP5 may play a role in RA pathogenesis. CD300LG is a novel O-glycosylated member of the CD300 antigen-like family. Besides a classical mucin-like domain, it contains a V-type Ig domain. CD300LG binds lymphocyte L-selectin via its Ig domain and supports lymphocyte rolling via its mucin-like domain. The unique structure and function of CD300LG suggest it may play an important role in inflammation [22].
These findings imply that the selected genes may contribute to or be a marker of the pathophysiology of RA treatment.
GO enrichment analyze.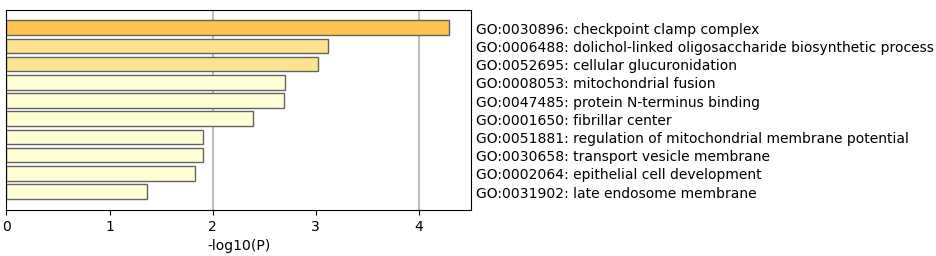
KEGG enrichment analyses.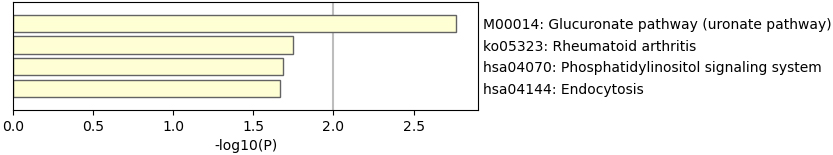
We then perform GO and KEGG enrichment analyses to the 69 genes, as shown in Figs 2 and 3. The results of GO analysis shows the selected 69 genes are involved in 74 significant pathways (with
The enriched pathways may role in RA treatment. It is becoming increasingly recognized that immune checkpoint inhibitors can result in inflammatory arthritis among patients treated with these drugs [23]. Checkpoint clamp complex pathway may play an important role in RA development. The genes in the cellular response to ionizing radiation may affect the effectiveness of anti-TNF therapy.
These pathways might offer a unique time-lapse window into the inflammatory arthritis process by which immune-related adverse events occur and predict or prevent them. They may also provide a unique window into the early occurrence of inflammatory arthritis in humans.
Table 1 and Figs 1–3 suggested that selected 69 genes might reveal the biological process of the treatment.
Discussion
A systemic inflammatory disease, RA is manifested by destructive distal polyarthritis. It can cause progressive joint damage, affect other organs, and even lead to cardiovascular disease unless diagnosed and treated. Targeted therapy using anti-TNF is the first option for patients with RA. Anti-TNF therapy, however, does not lead to meaningful clinical improvement in many RA patients. To predict which patients will not benefit from anti-TNF therapy, clinical tests should be performed prior to treatment beginning. Although various efforts have been made to identify biomarkers and pathways that may be helpful to predict the response to anti-TNF treatment, gaps remain in clinical use due to the low predictive power of the selected biomarkers. In this paper, we used a network-based computational method to identify the select the predictive biomarkers to guide the treatment of RA patients. We select 69 genes from peripheral blood expression data from 46 subjects using a sparse network-based method. The result shows that the selected 69 genes might influence biological processes and molecular functions related to the treatment.
Conclusion
Our approach advances the predictive power of anti-TNF therapy response and provides new genetic markers and pathways that may influence the treatment. One limitation of this paper is the lack of deep verification of the selected genes and network module.
Footnotes
Acknowledgments
This work was partially funded by the National Natural Science Foundation of China (62102261, 61976052), the Science and Technology Development Fund, Macau SAR (0056/2020/AFJ, 0158/2019/A3), the Jihua laboratory scientific project (X210101UZ210), the Foshan scientific project (2018AB003621), the School Moral Education Research project of Guangdong Education Department (2019GXSZ059), the Special Innovation Projects of Universities in Guangdong Province (2018KTSCX205), and the Science and Technology Project of Shaoguan City (200811104531028).
Conflict of interest
None to report.