
Abstract
BACKGROUND:
DNA sequence alignment is one of the most fundamental and important operation to identify which gene family may contain this sequence, pattern matching for DNA sequence has been a fundamental issue in biomedical engineering, biotechnology and health informatics.
OBJECTIVE:
To solve this problem, this study proposes an optimal multi pattern matching with wildcards for DNA sequence.
METHODS:
This proposed method packs the patterns and a sliding window of texts, and the window slides along the given packed text, matching against stored packed patterns.
RESULTS:
Three data sets are used to test the performance of the proposed algorithm, and the algorithm was seen to be more efficient than the competitors because its operation is close to machine language.
CONCLUSIONS:
Theoretical analysis and experimental results both demonstrate that the proposed method outperforms the state-of-the-art methods and is especially effective for the DNA sequence.
Introduction
In recent years, the biological data has significantly increased. DNA sequence is typical biological data that contains basic information of species [1, 2]. DNA sequencing is important in practical areas such as life and agricultural sciences. But nowadays, the DNA sequence data with massive size is a significant computational challenge [3]. Therefore, efficient DNA sequences processing is a major problem to cope with. Pattern matching between different species, i.e., pattern matching for DNA sequence has been a fundamental issue in biomedical engineering, biotechnology and health informatics.
In bioinformatics, DNA sequence alignment is one of the most fundamental and important operation to identify which gene family may contain this sequence. It can be inferred the biological function the sequence may have by DNA sequence alignment. The mechanism of DNA sequence alignment allows the target sequence to be compared with other sequences to find regions of local similarity [1], this process can be modelled as pattern matching problem, in which a pattern is a sequence of DNA characters (A, C, G, T) that is to be searched in a DNA sequence or chromosome [3].
Optimal multi pattern matching for DNA sequence is a challenging issue. For example, when we want to find the locational of biological virus in the genetic sequence, since the virus is not simply copied, some short sequence fragments may be inserted and deleted between two adjacent characters, in this case, pattern matching algorithm with wildcards is necessary [4].
In this paper, we propose a novel algorithm for optimal multiple pattern matching with wildcards for DNA sequence based on a packed string. The algorithm has a pre-processing time complexity of
The remainder of this paper is organized as follows: Section 2 provides the problem definition and notations. The proposed method is described in Section 3, and then a theoretical analysis of the method is outlined. The experimental results are presented in Section 4. Lastly, conclusions and future works are summarized in Section 5.
Definition and notations
The aim of this paper is to find the occurrence of each pattern from set
occ denotes the number of occurrences for pattern
Let
The main idea of the proposed algorithm is to pack each
Slide window on 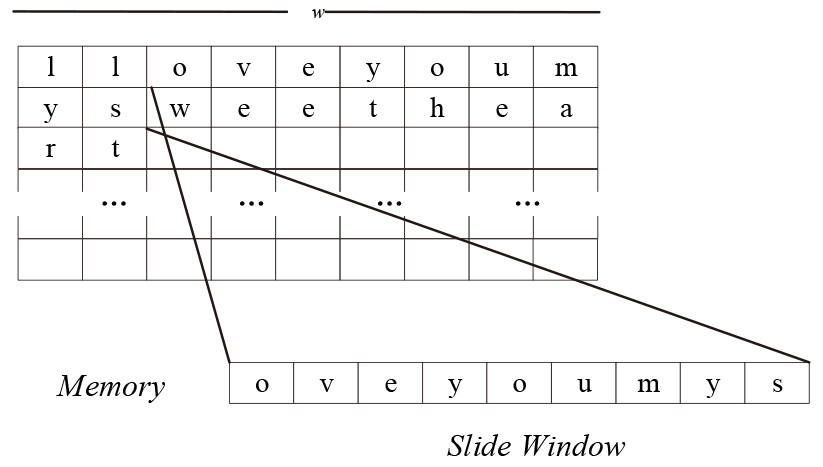
Assume that the machine word
The pre-processing phase is first executed to; encode every symbol in text and patterns with a unique integer number and wildcards with 0’s. All the pattern symbols are packed to machine words. Let us assume now that every pattern can be encoded to a single machine word, i.e.,
The result is stored as a packed string pattern,
The text
Comparison process
Comparison of a packed string in a sliding window with a packed string of stored patterns can be achieved as follows. Let us assume that the size of the sliding window and the size of each pattern are identical, and that the wildcard bits in a packed string of patterns are all 1’s and the wildcard bits in the text are all 0’s. Then, we have
The complement of the packed string of a pattern is first computed. Then, an AND bit-wise operation is executed between the packed string of the pattern and the sliding window, where the bits of wildcards are all 0’s, both in the sliding window and in the complement of the packed string of the pattern. A match exists for the pattern at position
The second process is called as double checking. In this process, the OR bit-wise operation is executed between the complement of a packed string of pattern and a packed string of text. During the OR-ing process, the wildcard bits should all be 1’s. If the bits of a resulting word are all 1’s, then the pattern
To check the match of the remaining part of the word if a pattern cannot be packed into a single machine word, i.e.,
A Pseudo Code of the Proposed Algorithm
Input a set of patterns Encode all the symbols in patterns and text with unique number and wildcards with 0. Pack each
OR the new character with shifting result. Pack the first
OR the new character with shifting result. Compare the packed string in the sliding window with the packed string of every patterns.
Change wildcards bits to 0’s for both the sliding window and the complement of pattern. AND the packed string of sliding window the complement of every patterns. If the result is 0.
Change the wildcard of the sliding window and the complement of patterns to all of 1. OR packed string of sliding window and the complement of every pattern. If the bits of the result of pattern Shift sliding window by
Assuming that pattern set
In each shifting time of updating the sliding window, only one character is added to the sliding window, so the time complexity of updating sliding window is
Table 1 shows a theoretical comparison between our algorithm and other five recently proposed algorithms. The comparison includes the algorithmic performance and support for matching with Long Pattern (LP), Big Alphabet size (BA) and Non-Interrupted Pattern Updating (NPU).
Comparison of algorithms solving the problem of pattern matching with fixed number of wildcards
Comparison of algorithms solving the problem of pattern matching with fixed number of wildcards
Three data sets were used to test the performance of the proposed algorithm. The first two data sets source from natural language data and the last one sources from a DNA sequence data set. The first nature language data set is from enwik8 (Wiki.) corpus that compromises the first 100 MB of Wikipedia in English language with an alphabet size of 127 different symbols. The second natural language data set is the 4.245 MB King James Version (KJV) of the Bible with an alphabet size of 79 different symbols. The third data set is 96.7 MB hs13.chr file corresponding to the plain ASCII form of the human chromosome 13 (DNA) and the alphabet size is 10 different symbols. Human chromosome 13 is selected because it has moderate size, and it is related to breast cancer, which makes it significant to be analyzed.
The experiments were conducted on a 2.16 GHz Pentium (R) dual 64-bit CPU computer with 2 G RAM and 32-bit Microsoft Windows 8 operating system; we utilized C++ language and Microsoft Visual Studio 2005 to implement the algorithms. We conducted the bench marking of the algorithms in a consistent and controlled environment, and the wildcard character was selected randomly.
The performance of the proposed algorithm was tested against
Pre-processing time for the proposed algorithm applied on three data sets.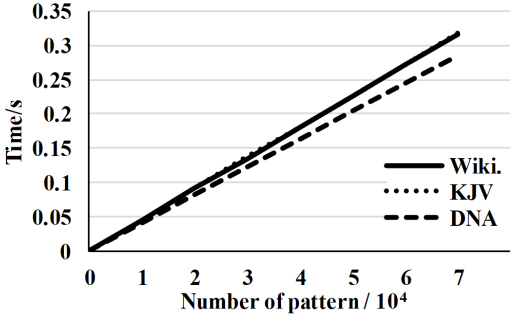
Performance of the proposed algorithm on three data sets.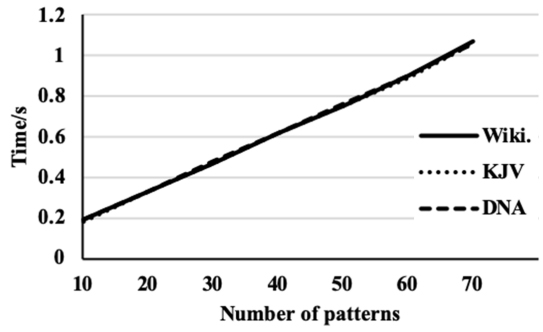
The proposed method ran efficiently in the pre-processing step on the three data sets. Figure 2 shows that for the natural language data set, the pre-processing time was almost identical for both the Wiki and KJV datasets, but it was less for the DNA data set. The figure also shows that the pre-processing time was linear as a function of the number of patterns for all three data sets. In a numerical form, the pre-processing time in sec. for the three data sets, the proposed algorithm can process one hundred thousand of patterns in less than half a second of where every pattern has 8 characters. This means that the algorithm can process almost one million characters in less than half a second.
Comparison of pre-processing on the DNA data set where 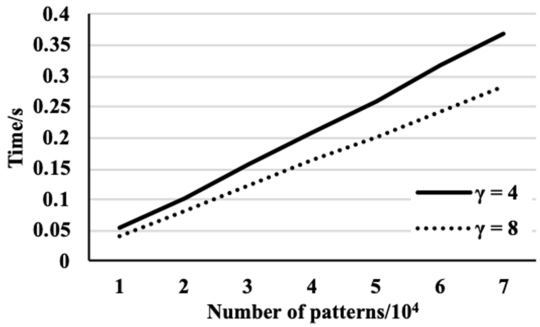
Figure 3 shows that the algorithm performance on the three data sets. From the figure it can be observed that the time complexity for the three data sets is identical and linear. The experimental results shown in Fig. 4 are achieved on the data set that has the smallest alphabet size, i.e., the DNA data set. The dataset is with 10 different symbols from an alphabet where
The time of pre-processing for three algorithms on the Wikipedia data set.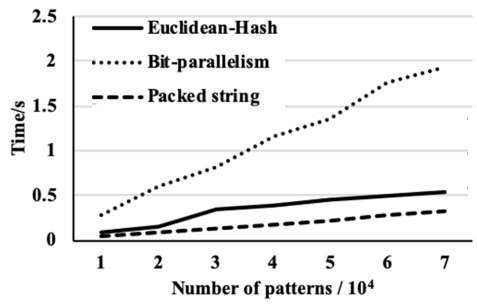
We then compare the performance of the proposed algorithm with the performance of two recent algorithms. The first one is based on the Euclidean distance and the hash function [5] and the second is based on bit-parallelism [6]. Figures 5–7 show the pre-processing time of the three algorithms against the number of patterns for the three data sets. The figures also show that the bit-parallelism based algorithm is less efficient in the pre-processing step than the other two algorithms where the packed string-based algorithm is the most efficient. While the pre-processing time of the packed string-based algorithm is close in pre-processing time with the Euclidean distance and the hash function-based algorithm.
The performance (time/s) of three algorithms on the Wikipedia data set
The performance (time/s) of three algorithms on the KJV data set
The time of pre-processing for three algorithms on the KJV data set.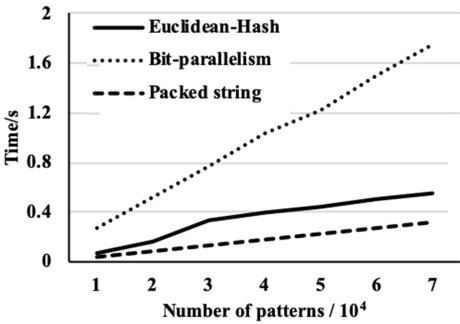
The time of pre-processing for three algorithms on the data set.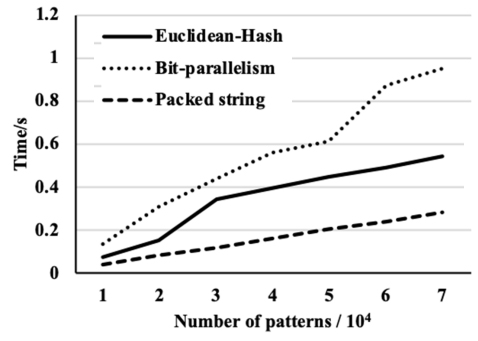
The performance (time/s) of three algorithms on the DNA data set
Tables 2–4 show the performance of the algorithms in seconds as a function of the number of patterns on the first 1 MB of the three data sets. As shown in the tables, for the three data sets, the bit-parallelism-based algorithm and the packed string-based algorithm demonstrate a close performance. Of the two, a better performance is observed for the packed string-based algorithm proposed in this paper. Again, it should be stressed that the Euclidean distance and hash function-based algorithm has less efficiency because it uses the Fast Fourier Transform (FFT) which has many interior variables which slows down the algorithm.
Pattern matching for DNA sequence has been an essential issue in biomedical engineering and health informatics. The main finding of this article is to propose a faster pattern matching for DNA sequence, i.e., a practical and efficient algorithm for multi-pattern matching with a fixed number of wildcards is proposed. The main idea of the proposed method is to consider matching patterns as packed string patterns with a sliding window. The window slides along the given packed string text, matching against stored packed string patterns. Three data sets are used to test the performance of the proposed algorithm and compare with the state-of-the-art methods. The proposed algorithm was seen to be more efficient than the competitors because its operation is close to machine language (the algorithm operates machine word, so CPU can directly interpret the data), the proposed algorithm is especially effective for the DNA sequence. In future work, the efficiency of algorithms can be improved by merging the proposed bit-parallelism and string packing algorithm with one or more mechanisms such as Euclidean distance and Hash function [7]. In particular, the proposed algorithm can be improved by using specialized word-size packed string-matching instructions. We also plan to improve the work by combining uncertain reasoning models such as evidence theory [8].
Footnotes
Conflict of interest
None to report.