
Abstract
While workflow systems have improved the repeatability of scientific experiments, the value of the processed (intermediate) data have been overlooked so far. In this paper, we argue that the intermediate data products of workflow executions should be seen as first-class objects that need to be curated and published. Not only will this be exploited to save time and resources needed when re-executing workflows, but more importantly, it will improve the reuse of data products by the same or peer scientists in the context of new hypotheses and experiments. To assist curator in annotating (intermediate) workflow data, we exploit in this work multiple sources of information, namely: (i) the provenance information captured by the workflow system, and (ii) domain annotations that are provided by tools registries, such as Bio.Tools. Furthermore, we show, on a concrete bioinformatics scenario, how summarising techniques can be used to reduce the machine-generated provenance information of such data products into concise human- and machine-readable annotations.
Introduction
We have witnessed in the last decade a paradigm shift in the way scientists conduct their experiments, which are increasingly data-driven. Given a hypothesis that the scientist seeks to test, verify or confirm, s/he processes given input datasets using an experiment which is modelled as a series of scripts written in languages such as R, Python and Perl, or pipelines composed of connected modules (also known as workflows [8,24]). For example, the recent progress in sequencing technologies, combined with the reduction of their cost has led to massive production of genomic data with growth rates that exceed major manufacturers’ expectations [30]. A single research lab that is using the last generation sequencer can currently generate in one year1
Theoretically around 2500 whole genomes per year with an Illumina NovaSeq technology.
The datasets obtained as a result of the experiment are analyzed by the scientist who then reports on the finding s/he obtained by analyzing the results [3]. As a response to the reproducibility movement [31], which has gained great momentum recently, scientists were encouraged to not only report on their findings, but also document the experiment (method) they used, the datasets they used as inputs, and eventually, the datasets obtained a result. To assist scientist in the task of reporting, a number of methods and tools have been proposed (see e.g., [4,12,17]). In [19] Gil et al. propose data narratives to automatically generate text to describe computational analyses that can be presented to users and ultimately included in papers or reports.
While we recognize that such proposals are of great help to the scientists and can be instrumental to a certain extent to check the repeatability of experiments, they are missing opportunities when it comes to the reuse of the intermediate data products that are generated by their experiments. Indeed, the focus in the reports generated by the scientist is put on their scientific findings, documenting the hypothesis and experiment they used, and in certain cases, the datasets obtained as a result of their experiment. The intermediate datasets, which are by-products of the internal steps of the experiment, are in most cases buried in the provenance of the experiment if not reported at all. The availability of such intermediate datasets can be of value to third-party scientists to run their own experiment. This does not only save time for those scientists in that they can use readily available datasets but also save time and resources since some intermediate datasets are generated using large-scale resource- and compute-intensive scripts or modules.
We argue that intermediate datasets generated by the steps of an experiment should be promoted as first-class objects on their own right, to be findable, accessible and ultimately reusable by the members of the scientific community. We focus, in this paper, on datasets that are generated by experiments that are specified and enacted using workflows. There has been recently initiatives, notably FAIR [34], which specify the guidelines and criteria that need to be met when sharing data in general. Meeting such criteria remains challenging, however.
In this paper, we show how we can combine provenance metadata with external knowledge associated with workflows and tools to promote processed data sharing and reuse. More specifically, we present
The contributions of this paper are the following:
Definition of workflow data products reuse in the bioinformatics domain.
A knowledge-graph based approach aimed at annotating raw processed data with domain-specific concepts, while limiting domain experts overwhelming at the time of sharing their data.
An experiment based on a real-life bioinformatics workflow, that can be reproduced through an interactive notebook.
This paper is organised as follows. Section 2 presents motivation and defines the problem statement. Section 3 details the proposed
We motivate our proposal through an exome-sequencing bioinformatics workflow. This workflow aims at (1) aligning sample exome data (the protein-coding region of genes) to a reference genome and (2) identifying genetic mutations for each of the biological samples. Figure 1 drafts a summary of the bioinformatics analysis tasks required to identify and annotate genetic variants from exome-sequencing data. For a matter of clarity, we hide in this scenario some of the minor processing steps such as the sorting of DNA bases, but they are still required in practice. The real workflow will be described in detail in the experimental results section.

A typical bioinformatics workflow aimed at identifying and annotating genomic variations from a reference genome. Green waved boxes represent data files, and blue rounded boxes represent processing steps.
This workflow consumes as inputs two sequenced biological samples
Performing these analyses in real-life conditions is computation intensive. They require a lot of CPU time and storage capacity. As an example, similar workflows are run in production in the CNRGH french national sequencing facility. For a typical exome-sequencing sample (9.7 GB compressed), it has been measured that 18.6 GB was necessary to store the input and output compressed data. In addition, 2 hours and 27 minutes were necessary to produced an annotated VCF variant file, taking advantage of parallelism in a dedicated high-performance computing infrastructure (7 nodes with 28 CPU Intel Broadwell cores each), which corresponds to 158 cumulative hours for a single sample, i.e. 6 days of computation on a single CPU.
Considering the computational cost of these analyses, we claim that the secondary use of data is critical to speed-up Research addressing similar or related topics. In this workflow, all processing steps produce data but they do not provide the same level of reusability. We tagged reusable data with a white star in Fig. 1. More precisely, (
From the scientist perspective, answering questions such as “can or should I reuse these files in the context of my research study” is still challenging. To reuse the final VCF variant file, it is of major importance to know the version of the reference genome as well as to clearly understand the scientific context of the study, the phenotypes associated to the samples, as well as the possible relations between samples. Finally, having precise information on the variant calling algorithm is also critical due to application-specific detection thresholds [26]. More generally, not only fine-grained provenance information regarding data and tools lineage are required but also domain-specific annotations based on community agreed vocabularies (Issue 1). These vocabularies exist but annotating processed data with domain-specific concepts requires a lot of time and expertise (Issue 2).
In this work, we show how we can improve the findability and reusability of workflow (intermediate) data by leveraging (1) community efforts aimed at semantically cataloguing bioinformatics processing tools to reduce the solicitation of domain experts, and (2) the automation and provenance capabilities of workflow management systems to automate the annotation of processed data, towards more reusable workflow results.

Knowledge graph based on workflow provenance and tool annotations to automate the production of human- and machine-oriented data summarises.
Being tightly coupled to scientific context, reusability is more challenging to achieve. Guidelines have been proposed for FAIR sharing of genomic data [14], however, proposing and evaluating reusability is still a challenging and work in progress [33]. In this work, we focus on reusable data as annotated with sufficiently complete information allowing, without needs for external resources: traceability, interpretability, understandability, and usage by humans or machines.
To be traceable, provenance traces are mandatory for tracking the data generation process.
To be interpretable, contextual data [34] are mandatory, this includes: (i) Scientific context in terms of Claims, Research lab, Experimental conditions, previous evidence (academic papers). (ii) The technical context in terms of material and methods, data sources, used software (algorithm, queries) and hardware.
To be understandable by itself, data must be annotated with domain-specific vocabularies. For instance, to capture knowledge associated with the data processing steps, we can rely on EDAM4
Figure 2 illustrates our approach to provide more reusable data. The first step consists in capturing provenance for all workflow runs. PROV5
is the de facto standard for describing and exchanging provenance graphs. Although capturing provenance can be easily managed in workflow engines, there is no systematic way to link a PROV Activity (the actual execution of a tool) to the relevant software Agent (i.e. the software responsible for the actual data processing). To address this issue we propose to provide, at workflow design time, the tool’s identifier in the tool catalogue. This allows to generate a provenance trace which associates (prov:wasAssociatedWith) each execution, and thus each consumed and produced data to the software identifier.Then, we assemble a bioinformatics knowledge graph which links together (1) the tools annotations, gathered from the Bio.Tools registry, and providing information on the functions of the tools (bioinformatics EDAM operations) and which kind of data they consume and produce, (2) the complete EDAM ontology, to gather for instance the community-agreed definitions and synonyms for bioinformatics concepts, (3) the PROV graph resulting from a workflow execution which provides fine-grained technical and domain-agnostic provenance metadata, and (4) the experimental context using Micro-publication for scientific claims and hypothesis associated to the experiment.
Finally, based on domain-specific provenance queries, the last step consists in extracting few and meaningful data from the knowledge graph, to provide scientist with more reusable intermediate or final results, and to provide machines findable and query-able data stories.
In the remainder of this section, we rely on the SPARQL query language to interact with the knowledge graph in terms of knowledge extraction and knowledge enrichment.

SPARQL query aimed at linking processed data to the processing tool and the definition of what is done on data.

SPARQL query aimed at assembling an abstract workflow based on what happened (provenance) and how data were processed (domain-specific EDAM annotations).
Query 1 aims at extracting and linking together data artefacts with the definition of the bioinformatics process they result from. In this SPARQL query, we first identify data (prov:Entity), the tool execution they result from (prov:wasGeneratedBy), and the used software (prov:wasAssociatedWith). Then we retrieve from the Bio.Tools sub-graph the EDAM annotation which specify the function of the tool (biotools:has_function). The definition of the function of the tool is retrieved from the EDAM ontology (oboInOwl:hasDefinition). Finally, we retrieve the scientific context of the experiment by matching statements expressed in natural language (mp:Claim, mp:statement).
Query 2 shows how a specific provenance pattern can be matched and reshaped to provide a summary of the main processing steps, in terms of domain-specific concepts. The idea consists in identifying all data derivation links (prov:wasDerivedFrom). From the identified data, the tool executions are then matched, as well as the corresponding software agents. Similarly, as in the previous query, the last piece of information to be identified is the functionality of the tools. This is done by exploiting the biotools:has_function predicate. Once this graph pattern is matched, a new graph is created using a CONSTRUCT query clause, to represent an ordered chain of processing steps (p-plan:wasPreceededBy).
Raw provenance traces from a bioinformatics workflow execution
We experimented our approach on a production-level exome-sequencing workflow,6
The resulting provenance graph consists in an RDF graph with 555 triples leveraging the PROV-O ontology. Table 1 and Table 2 show the distribution of PROV classes and properties respectively.
Number of instances per PROV class, resulting from the execution of the exome-sequencing workflow
Number of predicates per PROV and RDF(S) property, resulting from the execution of the exome-sequencing workflow
Interpreting this provenance graph is challenging from a human perspective due to the number of nodes and edges and, more importantly, due to the lack of domain-specific terms.
Based on query 1 and a textual template, we show in Fig. 3 sentences which have been automatically generated from the knowledge graph. They intend to provide scientists with self-explainable information on how data were produced, and in which scientific context, leveraging domain-specific terms.

Sentence-based data summaries providing, for a given file, information on the tool the data originates from, and the definition of what does the tool, based on the EDAM ontology.
The
Complex data analysis procedures would require a long text and many logical articulations for being understandable. Visual diagrams provide a compact representation for complex data processing and constitute thus an interesting mean to assemble human-oriented data summaries.
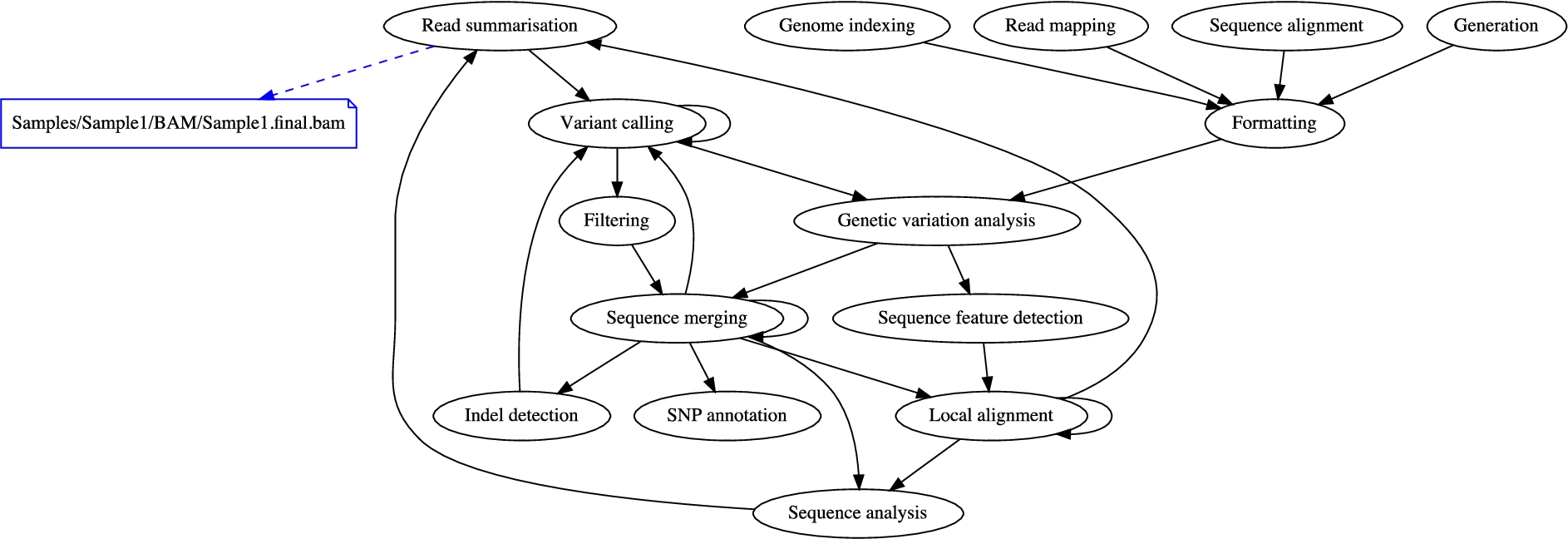
Fig. 4 shows a summary diagram automatically compiled from the bioinformatics knowledge graph previously described in Section 3. Black arrows represent the logical flow of data processing, and black ellipses represent the nature of data processing, in terms of EDAM operations. The diagram highlights in blue the

Human-oriented diagram automatically compiled from the provenance and domain-specific knowledge graph.
Another example for summary diagrams is provided in Fig. 5 which highlights the final VCF file and its binary index. The diagram shows that these files result from a processing step performing a SNP annotation, as defined in the EDAM ontology.
These visualisations provide scientists with means to situate an intermediate result, genomic sequences aligned to a reference genome (BAM file), or genomic variants (VCF file) in the context of a complex data analysis process. While an expert bioinformatician won’t need these summaries, we consider that visualizing and making explicit these summaries is of major interest to better reuse/repurpose scientific data, or even provide a first level of explanation in terms of domain-specific concepts.
Linked Data principles advocate the use of controlled vocabularies and ontologies to provide both human- and machine-readable knowledge. We show in Fig. 6 how domain-specific statements on data, typically their annotation with EDAM bioinformatics concepts, can be aggregated and shared between machines by leveraging the NanoPublication vocabulary. Published as Linked Data, these data summaries can be semantically indexed and searched, in line with the Findability of FAIR principles.

An extract of a machine-oriented NanoPublication aggregating domain-specific assertions, provenance and publication information.
Provenance capture. We slightly extended the Snakemake [21] workflow engine with a provenance capture module.7
Knowledge graph assembly. We developed a Python crawler8
Experimental setup. The results shown in Section 4 were obtained by running a Jupyter Notebook. RDF data loading and SPARQL query execution were achieved through the Python RDFlib library. Python string templates were used to assemble the NanoPublication while NetworkX, PyDot and GraphViz were used for basic graph visualisations.
We simulated the production-level exome-sequencing workflow to evaluate the computational cost of producing data summaries from an RDF knowledge graph. The simulation of the workflow execution allowed to not being impacted by the actual computing cost of performing raw genomic data analysis. Table 3 shows the cost using a 16 GB, 2.9 GHz Core i5 MacBook Pro desktop computer. We measured 22.7 s to load in memory the full knowledge graph (218 906 triples) covering the workflow claims and its provenance graph, the Bio.Tools RDF dump, and the EDAM ontology. The sentence-based data summaries have been obtained in 1.2 s, the machine-oriented NanoPublication has been generated in 61 ms, and finally 1.5 s to reshape and display the graph-based data summary. This overhead can be considered as negligible compared to the computing resources required to analyse exome-sequencing data as shown in Section 2.
Time for producing data summaries
To reproduce the human- and machine-oriented data summaries, this Jupyter Notebook is available through a source code repository.10
the overall data analysis process should be formalised into a computational workflow,
the running workflow management system should be able to dynamically capture generic provenance metadata as Linked Data, following the PROV-O ontology,
the run tools should be semantically annotated with domain-specific concepts. These descriptions should be accessible in a machine-actionable registry through a SPARQL endpoint,
mappings between workflow steps and the identifiers of the semantically annotated tools should be provided in the workflow specification so that provenance traces refer to semantically annotated tools.
The validation we reported has shown that it is possible to generate data summaries that provide valuable information about workflow data. In doing so, we focus on domain-specific annotations to promote the findability and reuse of data processed by scientific workflows with particular attention to genomics workflows. This is justified by the fact that
Enhancing reuse of processed data with
Regarding findability,
Regarding reusability, Table 4 points out the reusability aspects of
Still in the context of genomic data analysis, a typical reuse scenario would consists in exploiting as inputs, the annotated genomic variants (in blue in Fig. 4), to conduct a rare variant statistical analysis. If we consider that no semantics is attached to the names of files or tools, domain-agnostic provenance would fail in providing information on the nature of data processing. By looking on the human-oriented diagram, or by letting an algorithm query the machine-oriented nanopublication produced by
We focused in this work on the bioinformatics domain and leveraged Bio.Tools, a large-scale community effort aimed at semantically cataloguing available algorithms/tools. As soon as semantic tools catalogues are available for other domains,
In our work, we validated our solution by manually inspecting the usefulness of the summaries that are constructed given a real-life workflow. That said, we believe that there is a need for a benchmark that can be utilized by the community to systematically assess and compare the effectiveness of the proposed solutions. We also think that such a benchmark should be the result of a community-led effort to cater for different needs/requirements and different scientific domains.
Our work is related to proposals that seek to enable and facilitate the reproducibility and reuse of scientific artefacts and findings. We have seen recently the emergence of a number of solutions that assist scientists in the tasks of packaging resources that are necessary for preserving and reproducing their experiments. For example, OBI (Ontology for Biomedical Investigations) [9] and the ISA (Investigation, Study, Assay) model [27] are two widely used community models from the life science domain for describing experiments and investigations. OBI provides common terms, like investigations or experiments to describe investigations in the biomedical domain. It also allows the use of domain-specific vocabularies or ontologies to characterize experiment factors involved in the investigation. ISA on the other hand structures the descriptions about an investigation into three levels: Investigation, for describing the overall goals and means used in the experiment, study for documenting information about the subject under study and treatments that it may have undergone, and assay for representing the measurements performed on the subjects. Research Objects [5] is a workflow-friendly solution that provides a suite of ontologies that can be used for aggregating workflow specification together with its executions and annotations informing on the scientific hypothesis and other domain annotations. ReproZip [12] is another solution that helps users create relatively lightweight packages that include all the dependencies required to reproduce a workflow for experiments that are executed using scripts, in particular, Python scripts.
The above solutions are useful in that they help scientists package information they have about the experiment into a single container. However, they do not help scientists in actually annotating or reporting the findings of their experiments. In this respect, Alper et al. [4] and Gaignard et al. [17] developed solutions that provide the users by the means for deriving annotations for workflow results and for summarizing the provenance information provided by the workflow systems. Such summaries are utilized for reporting purposes.
While we recognize that such proposals are of great help to the scientists and can be instrumental to a certain extent to check the repeatability of experiments, they are missing opportunities when it comes to the reuse of the intermediate data products that are generated by their experiments. Indeed, the focus in the reports generated by the scientist is put on their scientific findings, documenting the hypothesis and experiment they used, and in certain cases, the datasets obtained as a result of their experiment. The intermediate datasets, which are by-products of the internal steps of the experiment, are in most cases buried in the provenance of the experiment if not reported at all. The availability of such intermediate datasets can be of value to third-party scientists to run their own experiment. This does not only save time for those scientists in that they can use readily available datasets but also save time and resources since some intermediate datasets are generated using large-scale resource- and compute-intensive scripts or modules.
Of particular interest to our work are the standards developed by the semantic web community for capturing provenance, notably the W3C PROV-O recommendation, and its workflow-oriented extensions, e.g., ProvONE,13
The proposal by Garijo and Gil [19] is perhaps the closest to ours in the sense that it focuses on data (as opposed to the experiment as a whole), and generate textual narratives from provenance information that is human-readable. The key idea of data narratives is to keep detailed provenance records of how an analysis was done, and to automatically generate human-readable description of those records that can be presented to users and ultimately included in papers or reports. The objective that we set out in this paper is different from that by Garijo and Gil in that we do not aim to generate narratives. Instead, we focus on annotating intermediate workflow data. The scientific communities have already investigated solutions for summarizing and reusing workflows (see e.g., [11,29]).
The solution proposed by Starlinger et al. [29] aims at identifying similarities between workflows. The authors exploit three sources of information, namely the labels used to describe the modules that compose the workflow, the structure (i.e., dataflow) of the workflow, and authorship information. In doing so, the authors do not tackle the problem that the human user faces when trying to understand a potentially complex workflow. Such a solution can be envisaged when the aim is to effectively search similar workflows in a repository given an initial input workflow. Our objective is different in that we aim to promote the reuse not only of workflows but also of the data products that the execution of such workflows produce, and we do so by leveraging summarisation techniques to produce human-friendly account on the data products.
Cerezo et al. [11] proposed a conceptual workflow model, close to end-user’s domain of expertise, aimed at enhancing the sharing and reuse of scientific workflows. These conceptual workflows are conceived at workflow design-time and are then semi-automatically refined into concrete executable workflows through a set of semantic transformations. Although our approach tackles reuse in data-driven sciences, we focus on the reuse of intermediate produced/consumed data whereas Cerezo et al. focus on the reuse of the data transformation process itself. In addition, our approach is bottom-up, based on workflow executions, and tends to limit the solicitation of domain experts, by leveraging already running semantically annotated tools catalogues.
It is worth noting that our work is complementary and compatible with the work by Garijo and Gil. In particular, the annotations and provenance summaries generated by the solution we propose can be used to feed the system developed by Garijo and Gil to generate more concise and informative narratives.
Our work is also related to the efforts of the scientific community to create open repositories for the publication of scientific data. For example, Figshare16
In this paper, we proposed
So far, we have focused in