
Abstract
Serious adverse events (serious AEs) following the therapeutic use of Botulinum Toxin Type A (BoNT-A) are infrequent. Children with pediatric spasticity often have comorbidities that can cloud causation around an adverse event (AE). If a serious AE occurs, clear documentation of information sharing and informed consent as well as the provider-patient relationship are critical to minimizing litigation risks. Reviewing the litigation that has occurred following BoNT-A for pediatric spasticity can offer insight into how providers’ perspectives regarding this intervention may differ from those of the public who might serve as jurists. This article offers suggestions for content sharing during the consent process to optimize patient understanding about potential adverse events.
Keywords
Introduction
Procedural and practice strategies for reducing the risk of adverse events (AEs) when injecting Botulinum Toxin Type A (BoNT-A) for pediatric spasticity management are well reviewed [1, 2, 3, 4]. The goal of this article is to review AE types, the current BoNT-A drug labels and to evaluate the “lessons to be learned” from United States (US) litigation associated with BoNT-A injections for the treatment of pediatric spasticity. These elements in turn, can serve as a guide for enhancing provider communication during shared decision making and informed consent.
Botox (Onabotulinumtoxin. A) Pediatric spasticity dosing and FDA approval [1, 2, 4, 9, 11, 16, 24, 25, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40]. In the 30 years that followed the initial FDA label, pediatric Cerebral Palsy/spasticity dosing was guided by dosing/outcome studies, practice descriptions from thought leaders, serious AEs leading to the FDA BBW, and publications from Practice Safety Reviews and Consensus Papers. Dosing based on the current FDA labels (as of April 2021) for pediatric CP/spasticity are seen on the right. Peak dosing mentioned in the literature was 40 U/kg [41].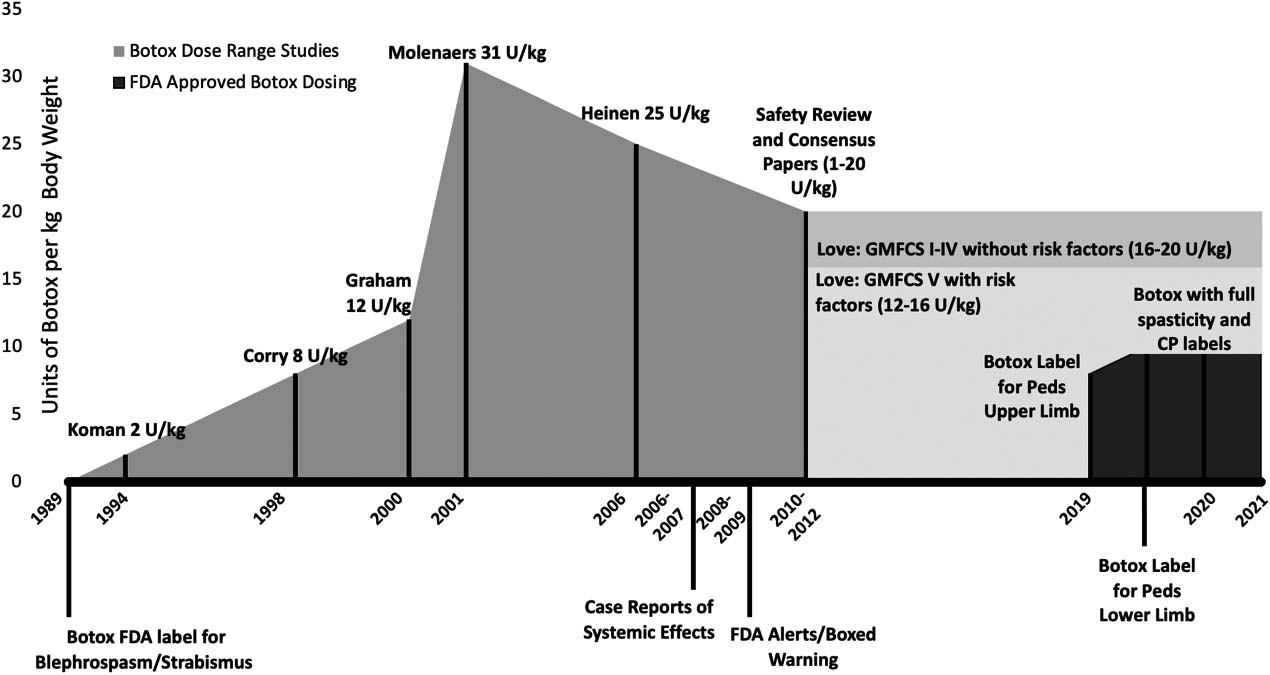
Dysport (AbobotulinumtoxinA). Pediatric spasticity dosing and FDA approval [1, 3, 4, 23, 26, 29, 32, 42, 43, 44, 45, 46]. The initial FDA label came after the BBW and dosing was guided by expert consensus. Dosing based on the current FDA labels (as of April 2021) for pediatric CP/spasticity are seen on the right. Peak dosing mentioned in the literature was 50 U/kg [13].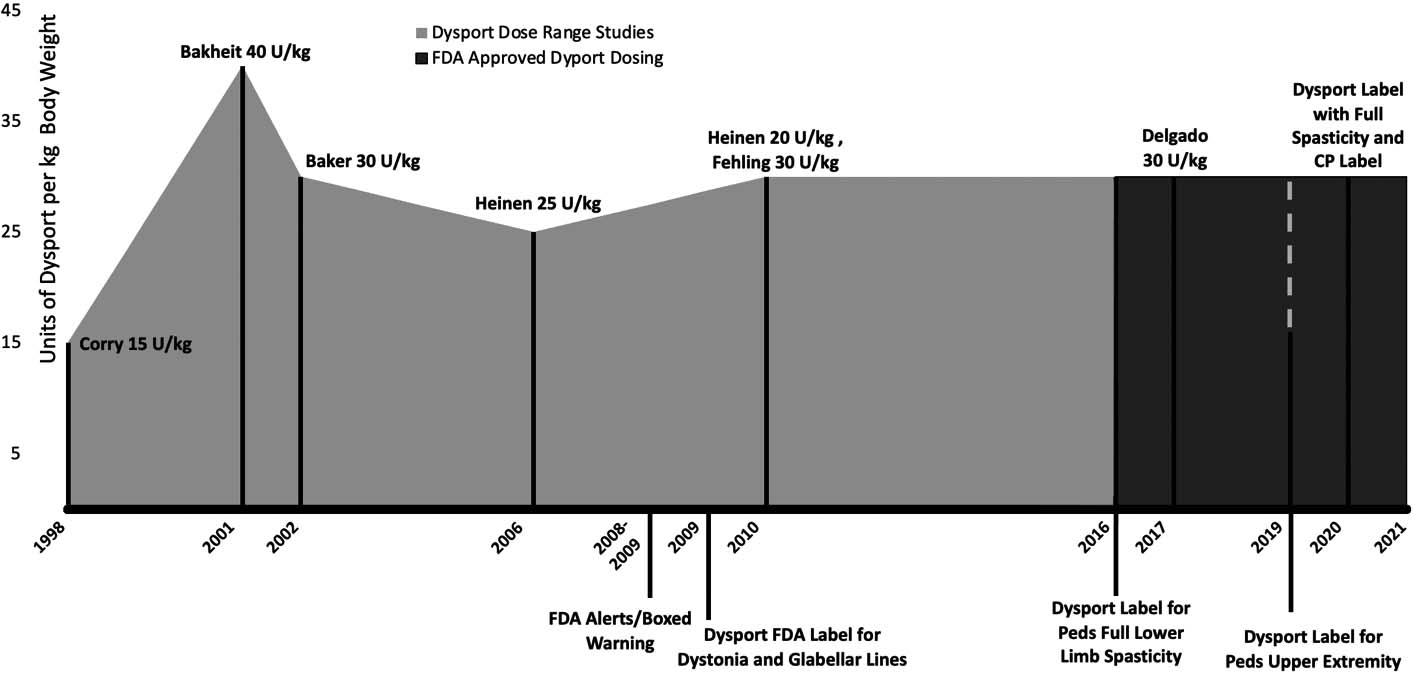
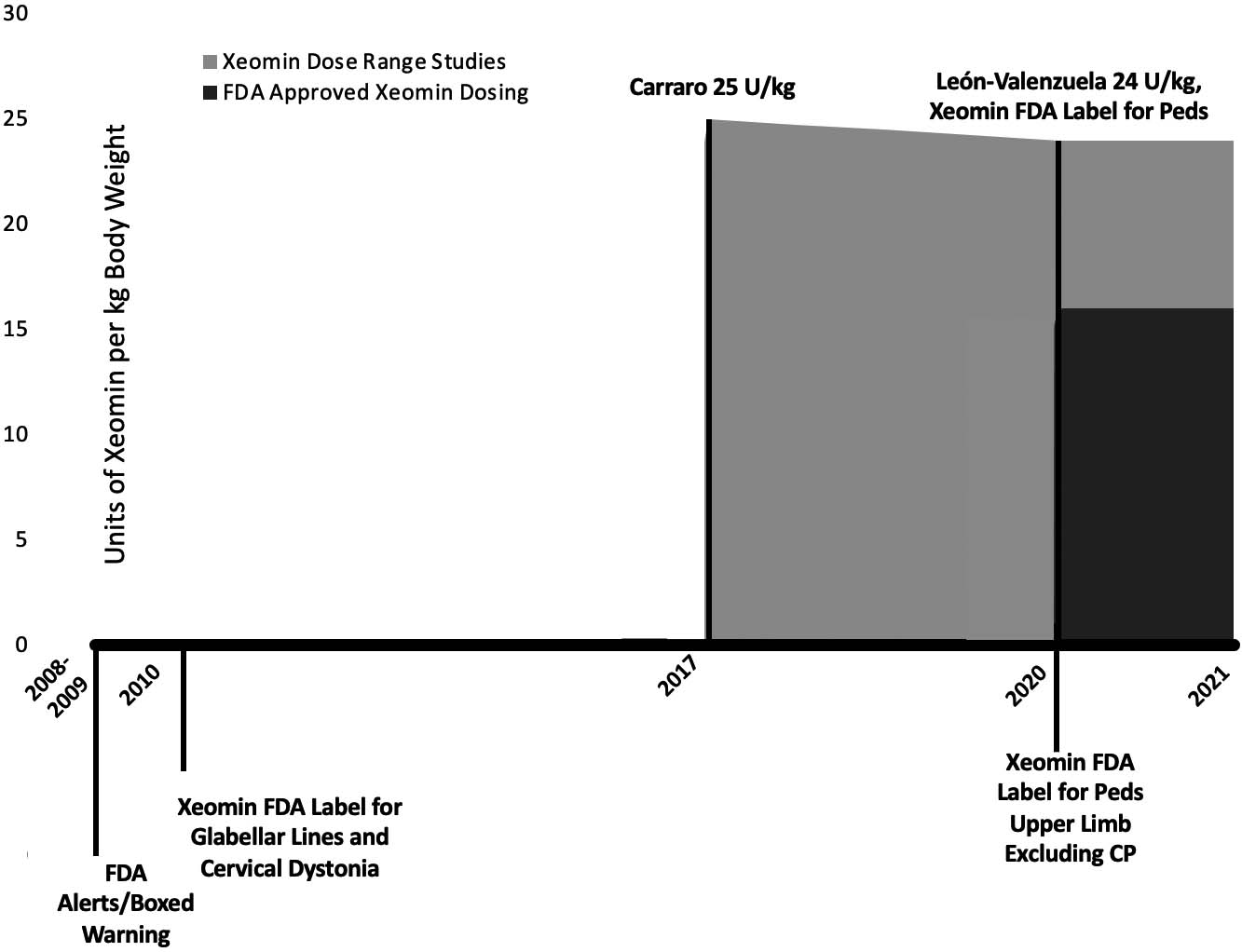
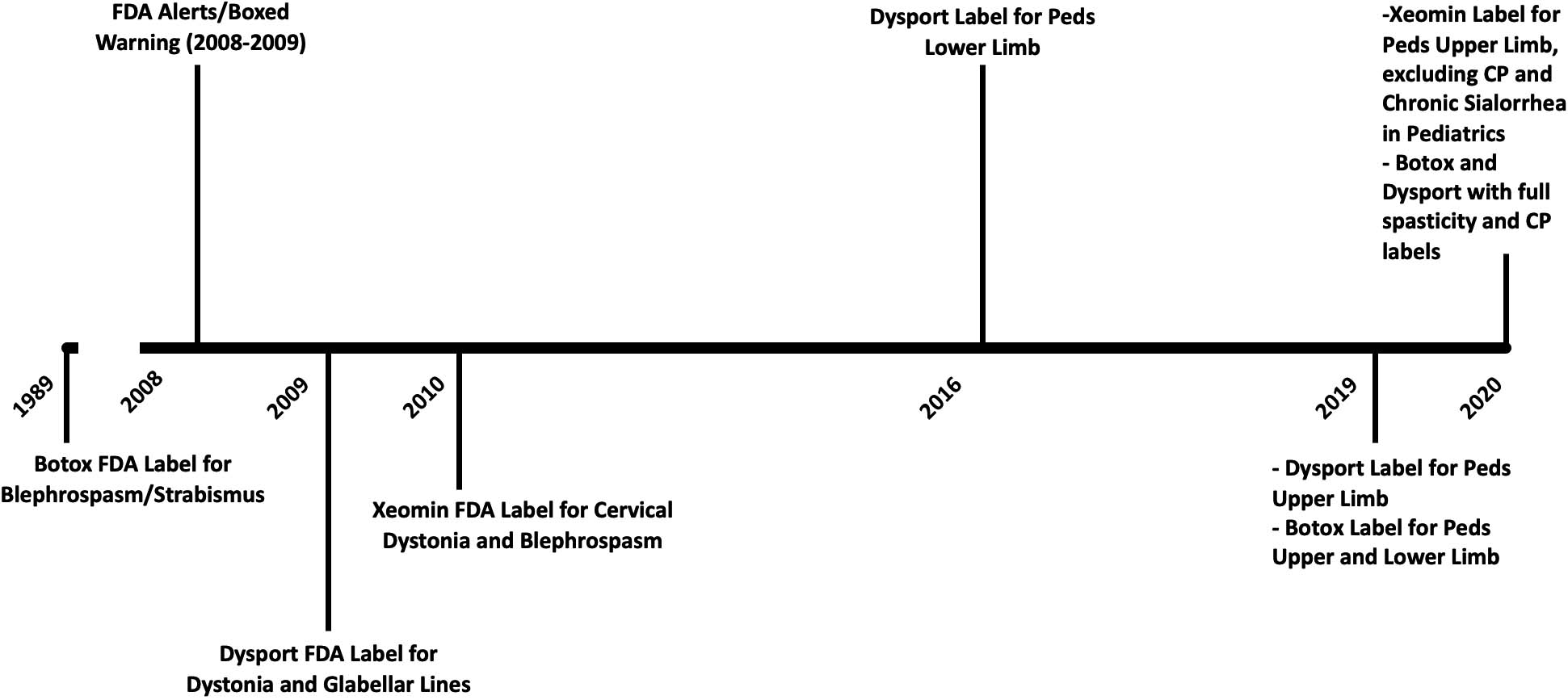
The timelines detailed in Figs 1–4 chronicle the published dosing studies, FDA warnings and indication approvals of BoNT-A products. Early medical literature around BoNT-A injections for pediatric spasticity focused on dosing, efficacy, technique and muscle distribution. In the decade preceding the Black Box Warning (BBW), there is a notable escalation in published doses (see Figs 2 and 3). In 2009, prior to the Federal Drug Administration’s (FDA) announcement of the BBW for all botulinum toxins, our pediatric spasticity literature began spotting infrequent cases of serious AEs. These were characterized by bulbar and generalized weakness, occurring with “higher doses” in more severely involved children [5, 6, 7]. In a purposed response to the rising safety concerns highlighted in these case descriptions and the BBW, the stakeholders in the injection community moved to publish results from several Practice Safety Reviews (2007–2010) [8, 9, 10, 11] and works from Expert Consensus Panels in 2010 [1, 2, 4]. Appropriately highlighted was the risk to children with higher Gross Motor Function Classification System (GMFCS) levels and in those children with comorbid dysphagia and aspiration [2, 9, 11, 12, 13, 14, 15, 16]. The medical literature has also recognized the baseline vulnerability of these children, in the absence of BoNT-A therapy [9, 12, 17]. This brings to light the potential for a temporally related event: a health incident (e.g., pneumonia, death) arising following a health intervention (including toxin injections) in a population at-risk. FDA reviews and adult based literature have also documented rare, idiosyncratic reactions [18, 19], and basic science studies [20, 21] have offered plausibility for these unusual reactions, including seizures.
UE – Upper extremity, LE – Lower extremity, NTE – Not to exceed, TBD – Total body dose.
The FDA drug label also known as the package insert is provided inside the packaging of all prescription medications. The label is the most complete information source of a prescription drug or biologic product. It is presented in a defined format and includes the necessary information for the healthcare professional to prescribe or counsel a medication. It covers all the FDA approved indications, AEs, and includes a boxed warning when one exists. Dosing for a specific indication in a drug’s label, is based on the outcomes data and safety findings from clinical trials submitted by clinician-scientists and the manufacturer to gain original FDA approval. Off-label drug use is defined as prescribing medications for indications, dosages, patient populations or dosage forms that have not received FDA approval [50]. Doctors are free to prescribe drugs for off-label use, but drug manufacturers may not promote off-label uses for their products [50]. They may however answer inquiries from providers regarding off-label use and refer them to peer-reviewed publications on the subject [50]. A drug label is updated when there is a newly approved indication or new safety information. Injectors should be aware of the current labels for all BoNT-A products used in their practice (Tables 1 and 2). The FDA does not regulate how a provider prescribes medications once they are on the market [50]. Off-label drug use is especially common in pediatrics and is well established in pediatric rehabilitation medicine [50, 51].
When does the FDA require a Medication Guide be given to the patient?
*This is our most common situation of performing an injection in a clinic setting.
After a drug product has been in use, if a new safety concern emerges, the FDA may determine that the labeled warning is inadequate by itself to provide for the safe use of a product. A boxed warning may be required. Whenever a drug has a boxed warning, the FDA requires the manufacturer produce a medication guide (MG), which is then included in the drug’s packaging. It may be physically separate from the package insert but it becomes part of a drug’s label. Typically a MG is provided to a patient when they pick up their prescription from the pharmacy. A MG is included inside every box of BoNT-A and is required by the FDA for all BoNT-A products because they meet two criteria: namely, certain information is necessary to prevent serious AEs, and patient decision-making might be altered based on information about these known serious AEs [53].
Injectors of BoNT-A serve two distinct roles as both providers and dispensers. This is an uncommon occurrence for providers who are more typically prescribers, rather than dispensers of medications. Dispensing is usually the role of a pharmacy or hospital. As previously noted, the FDA does not have jurisdiction over a provider’s practice, but it does when they serve as dispensers. The FDA states that you, the dispenser, must provide a BoNT-A product MG under certain conditions [53] (Table 3).
BoNT-A formulations are widely considered to have an excellent clinical safety profile. However, as with any treatment, they can produce AEs. Per the FDA an AE is “any untoward medical occurrence associated with the use of a drug in humans, whether or not considered drug related” [54]. The FDA definition of a serious AE, relevant to BoNT-A are events that result in:
death, or are life-threatening an inpatient hospitalization (such as pneumonia) or prolongation of existing hospitalization a persistent or significant incapacity or substantial disruption of the ability to conduct normal life functions other serious events such as bronchospasm or seizure not requiring hospitalization [54]
Most drug reaction types are Type A (dose-related) or less frequently, Type B (non-dose-related) [55]. Type A reactions are relatively common, predictable, related to the pharmacologic action of the drug, and have low mortality [55]. Management of this type of reaction focuses on reducing or discontinuing the therapy and considering interactions of the medication with other treatment modalities [55]. Type B reactions are uncommon, unpredictable, do not appear to be related to the pharmacological action of the drug, and have a high mortality [55]. Type B reactions can be further classified into immunologic, such as an allergic reaction, or idiosyncratic (as with abnormal liver function levels after initiation of treatment with dantrolene) [55, 56]. After a Type B reaction, the offending medication should be discontinued and avoided in the future [55]. Idiosyncratic reactions to BoNT have been reported in the literature after administration for a variety of treatment indications, including spasticity [19, 57].
Litigation involving BoNT-A used to treat pediatric spasticity
A recent analysis of BoNT related US litigation, published in 2013, found that lawsuits related to complications from BoNT products were uncommon, more likely from therapeutic than cosmetic use, and typically involving product liability claims against the manufacturer [58]. The greater number of therapeutic use related lawsuits likely reflects higher rates of AEs associated with higher dosing, comorbidities, polypharmacy [18], and off label injection of BoNT in children with muscle spasticity [58]. All the lawsuits involved onabotulinumtoxinA, probably associated with its longer duration on the market and its multiple indications. The review identified both patient-driven litigation over AEs and FDA legal action with Allergan, Inc. related to onabotulinumtoxinA. Allergan “agreed to plead guilty and pay $600 million to resolve its criminal and civil liability arising from the company’s unlawful promotion of its biological product Botox
What follows is a review of 3 cases of pediatric injection related litigation that have occurred in the US, since the BBW but prior to a labelled indication for pediatric spasticity. These cases had 3 distinct poor clinical outcomes following the injection of Botox product. Allergan was a defendant in each case. Providers’ involvement as co-defendants was case specific. Whether a provider was initially a codefendant or not, the reader should recognize that there is a significant emotional and practice burden for the providers throughout the litigation process, which can last for several years if it goes to trial. Efforts were made to identify other litigation involving other currently labeled BoNT-A products for pediatric spasticity, but no cases were found.
The first case resulted from an outcome of systemic botulism, the second from an idiosyncratic reaction and the last was a case of a temporally linked death that followed a series of injections. The purpose in presenting these cases is to gain understanding of the court’s perceptions, as they might differ from providers’ perspectives. This may inform clinical practices concerning the elements of communication with families that might be reviewed during the consent process. These case histories are drawn from court filings, so clinical details are scant. Rather than focus on the details that might satisfy a clinician’s interest, attention will be directed towards how a reasonable jury might perceive the adequacy of risk warnings the plaintiffs were provided.
Case #1
Case #1 was a preschool aged child with leukodystrophy resulting in lower limb spasticity who was injected with 200 units of Botox (18.33 U/kg). The case alleged that the Botox injections resulted in botulinum poisoning. After the injections, the child went on to require hospitalization, mechanical ventilation, and tube feeding. At the time of litigation, he continued to have decreased breathing capacity, muscle atrophy, and permanent weakness [61].
In this case, “Plaintiffs allege Defendant Allergan should be held strictly liable…for failure to warn about the risks of distant spread of toxin, botulism or botulism-like symptoms, and dose-related effects, meaning side effects are more likely to occur at higher doses.” [61]
Allergan argued that the learned intermediary doctrine applied and that the doctor acts as a learned intermediary between the patient and the prescription drug manufacturer by assessing the medical risks in light of the patient’s needs. The judge recorded “reasonable persons could differ on whether Allergan adequately warned (the provider) about the risk of toxin spread and botulism”. The judge noted that “despite having an internal maximum safe dose of 8 U/kg, Allergan responded” to the Doctor’s (previous) inquiry “by directing him to studies using up to 30 U/kg” [61]. Allergan, Inc. reached a confidential settlement during trial with the mother of this child [62].
Case #2
Case #2 was a 5.5-year-old male with GMFCS II diplegic cerebral palsy who was cognitively typically developing. At 3.5 years old he had a sub optimal response to Botox injections, approximately 6 u/kg total, into both calves. At 5.5 years he was re-injected with 12.33 u/kg total, 100 units in each calf. The next day he experienced facial swelling and reddening but it seemed to improve with Benadryl. The day after that his ears were also red. He vomited and different parts of his body began to twitch, including his eyes. He became unresponsive and was diagnosed as status epilepticus in the emergency room. He had another odd episode 3 months later that included red ears. 5 months later he was taken to the emergency room after he vomited. He was put on anti-seizure medications and diagnosed with epilepsy after an EEG revealed significant seizure activity. The injecting physician reported this as an AE to the Botox injection.
Almost 1.5 years following the injection, the patient’s parents filed a claim in court against Allergan, alleging that the seizure disorder was caused by the manufacturer’s negligence.
It was determined by the jury that Allergan was found to be negligent, and that the child suffered injuries as a result of that negligence. The jury awarded damages in favor of the plaintiffs.
Allergan subsequently filed a motion challenging the jury verdict and seeking a new trial. This required the judge to review the evidence and write an Opinion and Order regarding whether the jury, from the evidence presented, had been reasonable in reaching its conclusion. From this writing the judge and jury’s rationale may be understood, as it brings light to some of the details of testimony heard in deposition and at trial. As the jury had already returned a verdict finding liability, this Opinion and Order is written in a light most favorable to the plaintiffs. It does highlight risk elements that may help drive content for information sharing and consent.
To prove medical causation that Botox was the cause of (the child’s) seizure disorder and that negligence by Allergan caused (the doctor) to prescribe Botox in the dose selected, several pieces of evidence were provided to the court. Much of the evidence would be unfamiliar to treating clinicians: medical expert and Allergan leadership testimony, seizure rates reported in Allergan’s clinical trials, FDA correspondence including AE reports, corporate internal communications and evidence supporting theoretical mechanisms of BoNT spread. From the evidence, the Judge surmised, that a reasonable jury could conclude “that the Botox either caused the seizure outright or lowered the child’s seizure threshold to cause the seizure.” He supported the concept that “a preexisting condition that makes the plaintiff more susceptible to the event does not destroy causation” and that “reasonable jurors could have inferred that it was more likely than not that Botox was to blame for [the child’s] seizures.” Additionally, the jury could have reasonably concluded promotional activities were a substantial influence on the doctors dosing and practice [60, 63].
Case #3
The patient was a 21-year-old female with a history of microcephaly and cerebral palsy. Since the age of around 14 years old she had received several Botox injections for treatment of spasticity. The doses of Botox ranged from 10 U/kg to 17 U/kg. Throughout this time, the patient’s condition declined with multiple episodes of dysphagia following injections. One of these episodes required hospitalization for aspiration pneumonia. Her family stated that she lost use of her upper extremities 5 years after starting to receive the injections, and that her medical status unexpectedly declined during that timeframe. The patient continued to deteriorate throughout these 7 years, and she began having difficulty controlling her secretions. She also developed shallow breathing, difficulty with head control and seizures/seizure-like spells. The patient later died in her sleep and death was attributed to an atypical pneumonia.
The Plaintiffs in this case maintained that the weakness in the patient’s breathing, swallowing and gag reflex were a result of BoNT poisoning and that this weakness led to her death. While the patient was living, the Plaintiffs had inquired as to the cause of her decline, however Botox was never recognized to be related to these events by her medical team. The Plaintiffs also noted that they were unaware that the dosing the patient received was considered an overdose by Allergan’s standards, and this had only come to their attention after a news article was published regarding another Botox case (Case #2). They set to prove that Allergan promoted off-label dosing and failed to warn others about the safe dosing of Botox, the distant spread of the medication, and other side effect related Botox events [64]. Allergan reached a confidential settlement with the parents in this case [65].
Keeping our patients informed and our practices safe
These 3 cases illustrate litigation following a Type A reaction (botulism), a Type B reaction (seizure) and a temporally linked event (death). Conceivably any of these scenarios could occur in even the most conservative practice. When a serious AE occurs in practice, adequacy of consent and the Doctor-Patient Partnership are two critical elements in minimizing litigation risk that providers can control [66]. Because there is typically a time lapse between a negative outcome and testimony, a well-conceived and consistently delivered consent process affords the provider the confidence that appropriate information was shared. “I know I did this because this is what I always do” strongly supports the existing written consent in the medical record. The information shared with families related to consent should reflect the standard of shared decision making as cited in 2012, in Virtual Mentor, a publication of the AMA Journal of Ethics [67].
“Patients must have adequate information if they are to play a significant role in making decisions that reflect their own values and preferences, and physicians play a key role as educators in this process.” Providers “must disclose information that a reasonable person (or juror; mine) would want to have for decision making, even though that information may cause the patient to refuse treatment that the physician believes is in the patient’s best interest.”
More specific to this discussion is the following recommendation from the Canadian consensus group.
“The panel recommends routinely providing standardized verbal and written parental information about possible AEs of BoNT-A
Lessons learned: How might these cases influence the content of information sharing and consent
A. In the US, provision of the medication guide is the standard for communication when a drug product has a BBW.
B. Dosing above the label may be considered “an overdose” and failure to share this information during consent may influence a jury’s decision.
Prior to 2016, in the US all BoNT-A dosing for pediatric spasticity was off label. It was only in 2020 that labels were updated so that all 3 products now have indications for pediatric spasticity. Now that these indications exist it is conceivable that liability risks related to dosing might shift to providers when a serious AE occurs. Providers should understand their dosing practice as it relates to the label of each product used in their practice. A provider’s dosing decisions are driven by their training, experience, details of the child being injected and their risk tolerance. There are standards of care set forth in practice groups and in the medical literature. A drug label provides dosing guidance based on the clinical trials performed by the drug manufacturer to gain initial FDA approval for a specific indication. Where these various dosing standards are in alignment, dosing should not specifically need to be addressed during consent. When dosing exceeds the label, additional information sharing that includes reference to the literature and a provider’s experience may be prudent. As discussed, off-label drug practice is widely accepted and currently there is no legal precedence that states a physician must provide information that a medication’s use is off-label when consent is obtained [28]. It is therefore tenable that there is no need to be concerned with the disclosure of off-label BoNT-A dosing. Providing consent for dosing off-label, does however, bolster shared decision making and minimize litigation risks if an AE were to occur [58]. Keep in mind that during trial, the characterization of BoNT-A for the jury is that it is a deadly neurotoxin that carries a BBW. Additionally, if dosing above the label, it may be characterized as an “overdose” and that children with spasticity may be considered a vulnerable population. During the second case cited above the treating provider was asked in deposition, ‘If you’re planning on exceeding the maximum safe dose with a medication that’s a lethal neurotoxin, would you at least let the parents know about it so that they can consent to that?”
C. Regarding idiosyncratic reactions and seizures:
The second case illustrates that even if a serious AE is not likely a dose related reaction, the concept of an “overdose” may be deemed relevant by a jury.
Most injectors with a longer practice experience have encountered an AE of a change in seizure status following a BoNT-A injection. Some have also experienced a positive retrial following a subsequent injection, which leads them to no longer consider the treatment. It is appropriate to endorse this and other rare idiosyncratic events during consent, acknowledging the uncertainty regarding causation vs natural history and a temporal association for this population. This is an opportunity to review the risk of seizures inherent to any brain injury.
Minimizing litigation risks: Information sharing and the process of consent/assent
Providers are encouraged to stay risk aware. Two of the three litigation cases above occurred in an environment of another “newsworthy” BoNT-A case in the same community. If you, as a provider, have heard about a case in the news, so have patients and their families. Recognize that an experienced injector’s perception, as someone who has trained extensively and performed hundreds or thousands of injections, is quite skewed relative to a “jury of your peers.” A jurist likely only has cosmetic, or news related awareness of BoNT-A. Their understanding of this intervention will largely be shaped from their courtroom exposure.
Major mitigating factors in litigation risks after a negative outcome are the doctor-patient relationship and informed consent. When the risks, benefits, and alternatives to BoNT-A have been documented and completely discussed, it is a critical deterrent to negligence claims, making it difficult to lodge a complaint [66]. Reaching a decision to treat with BoNT-A is best if there is a collaborative partnership. BoNT-A injection is an effective yet elective, non-curative, treatment option that comes from a paralytic disease-causing bacteria with a name that ends in “toxin” and it carries the FDA’s most serious drug warning. Even when we believe it to be in the best interest of the patient, we must hold this perspective loosely. In this way, we position ourselves to respect a family’s preferences and risk tolerance. Recall that this is the purpose of the BBW, “that patient decision-making might be altered based on information about a known serious side effect” [53].
A hope for this article is the provision reflective insights that offer well “dosed” information sharing and prudent suggestions for practice risk reduction without transmitting undue fear to families. Towards that end, during initial patient encounters, be an educator. Discuss tone and tone reduction broadly including treatment options that are focal and systemic, reversible and irreversible. If goals are not purely focal, begin with less invasive, therapy interventions and oral medication. A brief delay in the use of BoNT-A offers a natural opportunity to build relationship, trust and refine goals before introducing this more invasive treatment. Children with only focal tone reduction goals typically receive lower dosing [13, 34, 46, 47] and tend to have lower GMFCS levels. As a result, they have a reduced risk of serious AEs, so earlier use of injections is rational.
We endorse appropriate provision of the MG and specific communication regarding its content, relevant focal weakness and seizures along with a discussion of dosing when off label. The pediatric BoNT-A literature encourages proactive education about serious AEs and intentionality in the seeking of AEs following injections [9, 11, 14, 16, 68]. Actively seeking AEs will raise a family’s awareness of possible AEs. This increases the likelihood of reporting, decreasing the risk of missing these events. The incidence of serious AEs is low but is more often observed in children with higher GMFCS levels [2, 11, 14, 16, 33], with attendant comorbidities and in those treated with higher doses [11, 42]. Our experience is consistent with Blaszczyk; caregivers very infrequently decline treatment after being informed about possible side effects [68]. Having shared goals and understanding of alternatives commonly balances a family’s reservations regarding risk. If a family is hesitant, their perspective will frequently evolve, especially if alternative interventions have met with limited treatment success or non-intervention has met with negative sequela.
The consent as a process
Pre injection visit
Prior to the procedure visit, it is important to establish goals for the initial injection with the family. Ideally these goals allow the first injection’s dose to be within the drug label. Families need the opportunity to digest the difficult to consider, contents of the BBW without an additional consideration of an “overdose”. Recognize that some families may require time or never be able to accept the risks expressed in the BBW. This ability to choose for/against a treatment option is a key principle of shared decision making.
Families should be provided an Information Sheet (IS) as well as the drugs MG on the visit prior to the injection. Covering serious AEs in this interaction before the day of the procedure allows a parent/adolescent to consider the risks and benefits and share this information with other family members. This discussion should also raise providers’ and parents’ mutual awareness of any recent health events, such as dysphagia or recent pneumonias, which might caution against BoNT-A therapy [16]. The IS, as part of the consent process, is an opportunity to share with families, in writing, information to guide decision making and should include a discussion of risks and benefits and a section on “reasons to delay injections” As this is an elective procedure, educate families that it is appropriate to postpone injections in the setting of significant respiratory illnesses or recent immunization that may carry its own rare risk of subsequent weakness (e.g., Guillain Barre Syndrome). Seizure instability is another reason to defer. Finally educate families about all BoNT product names. Be mindful of and coordinate with other treating providers also injecting BoNT for indications such as headache and sialorrhea. Avoid use of multiple BoNT products in the same time interval. This may be a particular risk as adolescents are transitioning to adult care.
Consent on the day of the procedure then becomes a review rather than an introduction of risk content, assuming no new family member accompanies the child. This documented, two-step delivery of risk information provides two benefits. It helps avoid day of procedure cancelations for health reasons or fears around risk awareness and provides additional assurance that written and verbal information sharing has occurred. Remember if a negative outcome results in litigation, testimony will likely occur one or more years later. If your standard of care is to extend risk discussion ahead of the injection, you will have complete confidence that these risks were discussed prior to the injection.
At the time of injection
Provide both written and verbal consent/assent that includes core content in every case and tailored content based on goals, dose, health and functional status. In addition to sedation and procedural risks, all serious AEs discussed in the BBW and the literature along with those in the injectors’ experience are appropriate for sharing. Communicate risks of muscle weakness: local, bulbar and systemic. Language communicating the severity of weakness such as the strength loss that could makes it difficult to breathe, clear secretions or swallow and can even become life threatening is necessary. Discuss idiosyncratic reactions, especially seizures, acknowledging uncertainty about temporal association versus an actual trigger in an at-risk child. If you have a specific case that allows you to discuss how you had to carefully consider future injections with the family, share that information generally. It helps families understand that serious AEs are rare but do occur and that our understanding of why they happen is incomplete. The MG can be provided again as a reference guide for the signs and symptoms of bulbar weakness. In a GMFCS IV or V child who develops a botulism-like illness, bulbar symptoms may be more identifiable than systemic weakness.
Discuss consequential focal weakness related to local spread, relevant to the muscles being injected and based on the child’s functional level. Dysphagia, incontinence and grip weakness may be germane, as they may meaningfully impact ADLs.
If dosing above the label per muscle, per limb and especially if exceeding the label’s total body dose, stay within consensus guidelines. Have additional discussions with the family that the dose being used is appropriate to the goals selected and that it is well established in clinical practice and medical literature. Acknowledge that rare case reports and practice safety reviews recognize that risks are higher when using higher doses and in children with higher GMFCS levels.
Risk dialogue around BoNT-A injections in GMFCS V children is an opportunity for a broader conversation of the child’s overall health related risks and life expectancy. Recent evaluations of this population highlight the shortened life expectancy associated with lower levels of mobility, limited capacity for oral feeding and the presence of epilepsy [12, 17]. One of our roles as educators is to help families understand these disability related risks. Risk communication around BoNT-A injections and other procedures can serve to prompt that extended conversation.
The need to dose above consensus guidelines should be infrequent, prompting consideration of alternative interventions. If undertaken they should be with thorough and well documented rationale and communication of risks.
Post injection
At follow up, in addition to a review of the benefits and goal attainment, proactively explore for any AEs with the BoNT-A procedure. Blaszczyk and others have used active surveillance of AEs through structured questionnaires [9, 13, 14, 68]. By actively seeking AEs in a written or verbal questionnaire, a more complete awareness of these events in practice will be gained, advancing the communication of risks to families [14]. At a minimum, inquiry and documentation regarding unexpected illness/hospitalizations, weakness, dysphagia and change in seizure status, should be explored (Supplement S1).
Review shared goals and then prior to a subsequent injection visit, if indicated, introduce the idea of dose escalation beyond the label (an overdose in the eyes of a jury). For Botox product especially, we do have ample supportive literature that can be referenced in this discussion. Raising this question after a well-tolerated BoNT-A event, while dosing on label, makes this risk consideration less overwhelming to a family. In our experience, families rarely reject off label dosing explained in this way.
Conclusions
Treatment with BoNT-A is an effective intervention for hypertonia caused by a variety of pediatric conditions. Consistent quality communication about this treatment, both written and verbal, is the optimal way to meet the information needs of families, facilitating their best treatment decision for their unique circumstances. Outlining potential risks and benefits in patient and family friendly language will optimize information sharing. An example is offered in Supplement S2. For BoNT-A, the relationship between serious AEs and CP severity is complex. In Swinney’s article, we find an opportunity for providing reasonable estimates of AEs and serious AE risks based on a child’s functional level [14]. This supplementary information can be provided to families who demonstrate a need for deeper inquiry; Supplement S3 is a sample Supplementary Information Sheet. Ideally, safety data would be sufficiently granular to identify specific pre- and post-injection rates of relevant AEs based on the elements of dosing, distribution, type of sedation, and patient features such as GMFCS level or associated conditions. In the absence of this detailed data, acknowledge these limits of our understanding and strive to inform families regarding the occurrence of rare serious AEs including the BBW, idiosyncratic and temporally associated AEs. The consent form offered in Supplement S4 is one such effort. This conversation should be contextualized to the specific child’s circumstance. Communication around BoNT-A treatment should not only describe and document treatment risks but is also an opportunity to improve a family’s understanding of their child’s health condition. Extending family knowledge in this way builds trust, promotes better shared decision making, and facilitates the Doctor-Patient relationship, keeping our patients and our practices safe.
Footnotes
Acknowledgments
The authors would like to thank James Banfield J.D., B.S. for his review of this manuscript and counsel, Deb Gaebler M.D. for her leadership, contributions and encouragement around this topic, and Darcy Fehlings M.D. for her insights and for sharing her consent for adaptation.
Conflict of interest
Ed Wright M.D., employed by Baylor College of Medicine and Speaker Honorarium from Hanger Inc. Lauren Fetsko D.O., employed by Baylor College of Medicine.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/PRM-210031.