
Abstract
Choledochal cysts (CC) are congenital biliary tract dilatations. Infantile CC (IFCC) in very low birth weight (VLBW) infants is rare. This is a case of a huge IFCC presented in VLBW preterm infant managed with external biliary drainage prior to definitive treatment. Electrolyte imbalance, poor weight gain, and infections were managed during external biliary drainage maintenance. Choledochal cyst excision and Roux-en-Y hepaticoenterostomy were successfully performed when the infant weighed 4.9 kg 5 months later. Delayed definitive treatment with external biliary drainage could be a feasible alternative for managing CC in low-birth-weight infants.
Abbreviations
Choledochal cyst
Very low birth weight
Introduction
Choledochal cyst (CC) is a rare congenital cystic biliary tract dilation. Although benign, CC can be associated with complications, including cholangitis, cholelithiasis, pancreatitis, and malignant transformation. Most CC cases warrant resection to avoid complications and future malignancies [1]. Using external biliary drainage before definitive surgery delays liver fibrosis progression by relieving biliary obstruction. This interim measure facilitates easier cyst dissection during the final repair [2]. Postponing the definitive surgical intervention for CC in high-risk infants could be considered to reduce the complications linked with early surgery.
Case report
A 29 + 1 weeks’, 1135 gm, very-low-birth-weight (VLBW) female infant was born to a 34-year-old, gravida 1, para 1 mother by vaginal delivery. Prenatal sonography and prenatal laboratory tests were normal. Antenatal corticosteroids were given once prior to delivery. After birth, the infant was intubated due to bradycardia and poor muscle tone. Apgar scores were 7 and 8 at 1 and 5 min, respectively. The infant was admitted to our neonatal intensive care unit.
The clinical course during the initial few weeks was uneventful. She was extubated and received noninvasive positive pressure ventilation on day of life (DOL) 8. On DOL 13, frequent bradycardia and cyanosis episodes were noted. No vomiting or acholic stools were observed. Chest X-ray revealed an abnormal density of the lower liver edge, raising suspicions of a mass lesion (Fig. 1a). Abdominal ultrasound demonstrated one cystic lesion connected with the gallbladder at the portal area, which was 4.5×4.0×3.1 cm, indicating type I CC (Fig. 1b). Laboratory data indicated hyperbilirubinemia. Considering the large size of the CC and the possibility of spontaneous rupture, surgery was necessary. However, complete excision of the cyst with bilioenteric anastomosis was not feasible in the VLBW infant (body weight: 1175 gm). Therefore, the choledochal cyst was managed by performing laparoscopic choledochostomy with external biliary drainage using an 8 Fr. pigtail catheter on DOL 14.
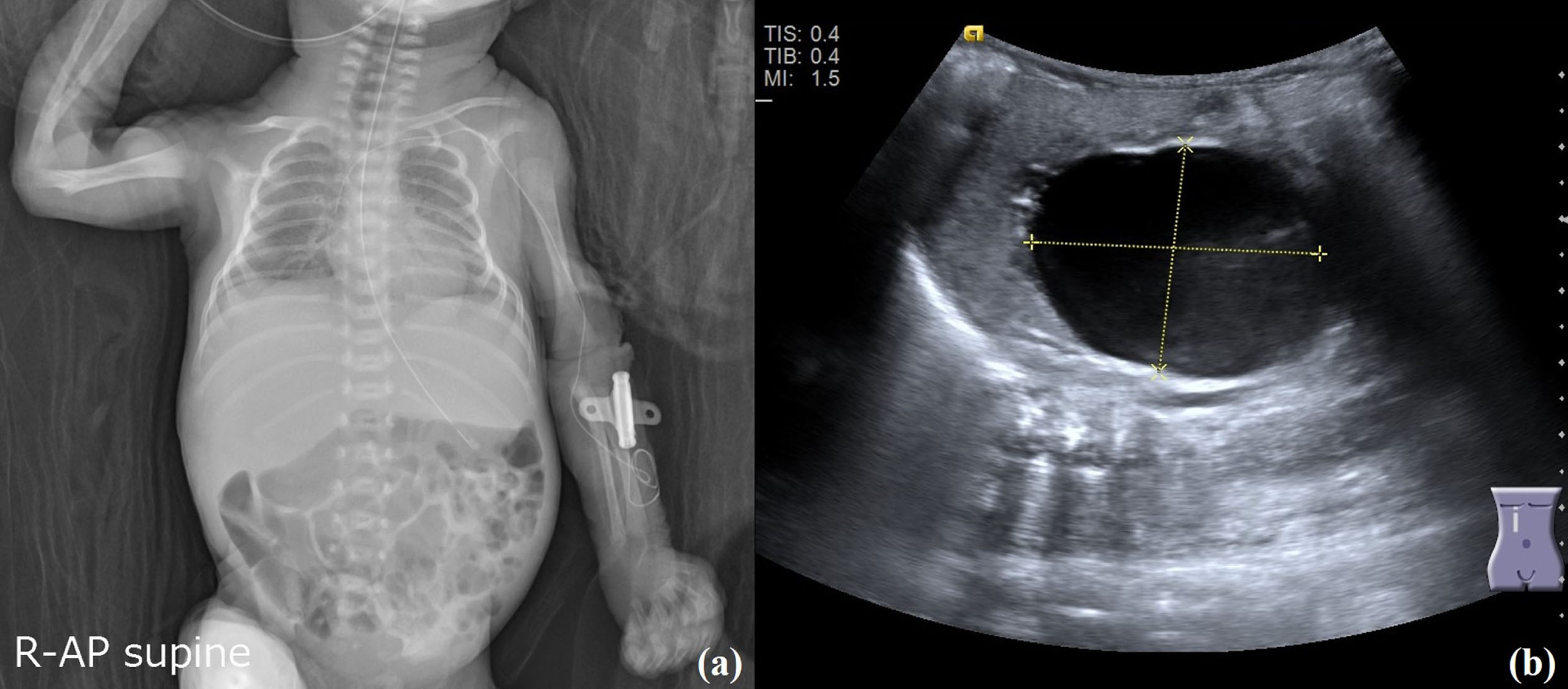
(a) Chest X-ray on DOL 13 with equivocal liver density. Abdominal ultrasound was performed according to this image finding and CC was diagnosed. (b) Abdominal ultrasound demonstrated one cystic lesion connected with the gallbladder at the portal area, 4.5×4.0×3.1 cm, suggesting type I CC.
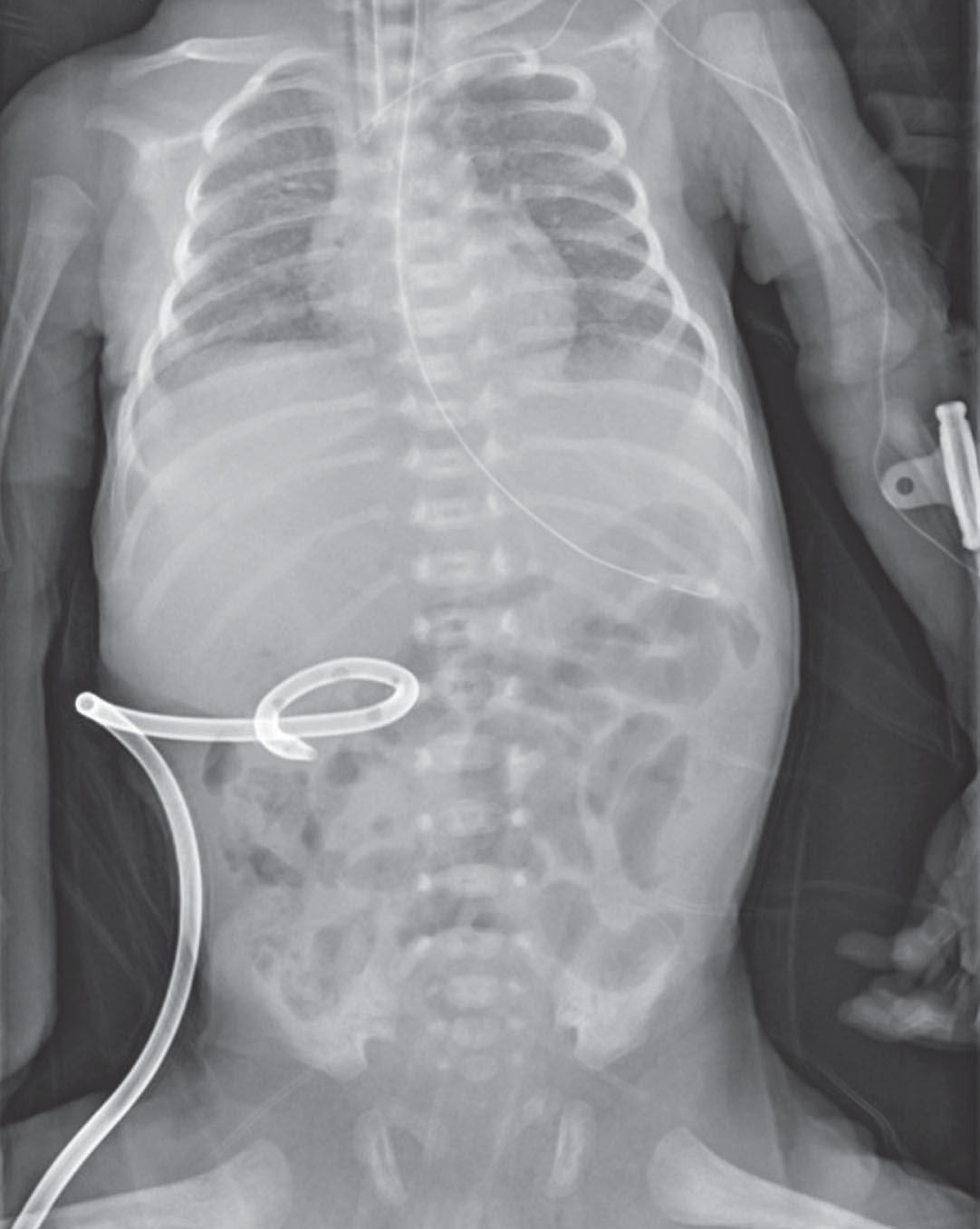
Chest X-ray showing a pigtail catheter for external biliary drainage after laparoscopic choledochostomy on DOL14.
Following the surgery, enteral feeding and parenteral nutrition were started, but electrolyte imbalance and poor weight gain with large amount of bile drainage were observed (25–40 ml/kg/day). We administered high-calorie premature infant formula containing maltodextrin and medium-chain triglyceride oil to address inadequate weight gain. The drainage loss was compensated by adjusting parenteral nutrition and recycling the bile via oral–gastric tube. Initially, frequent bradycardia and fluctuating O2 saturation were observed due to biliary reflux; therefore, we reduced the drainage amount by three times. Additionally, bile amount was titrated and the infant could tolerance that level. No abdominal fullness, mucus or blood in the stool, or steatorrhea was observed. Parenteral nutrition was discontinued due to an episode of sepsis. She was receiving enteral feeding (150 ml/kg/day), but the weight gain was slow due to persistent large bile drainage volume (30 ml/kg/day) and easy gastroesophageal reflux. A foul odor was noted from the biliary drainage on DOL 125, which contained Enterobacter aerogenes, Citrobacter koseri, and Enterococcus faecalis. We administered oral antibiotics including trimethoprim/sulfamethoxazole and ampicillin/sulbactam based on antibiotic sensitivity observed in the bile culture. This treatment was continued for 3 weeks until no microbes were isolated in subsequent bile cultures.
Laparoscopic CC excision and Roux-en-Y hepaticoenterostomy were successfully performed on DOL 168 when she weighed 4.9 kg. She experienced mild pancreatic leakage, which was resolved after one week of fasting and antibiotic administration. Feeding was restarted, which progressed smoothly. She was discharged with no oxygen, gavage, and full-volume enteral feeding on DOL 190 and a corrected GA of 3 months.
CC is a congenital biliary tract dilatation that is primarily seen in children. The incidence rate of CC is higher in Asian populations (1 in 13,000) than in Western populations (1 in 100,000). Approximately 80% CC cases are diagnosed during childhood and within the first decade of life [3]. Recent reports of CC in VLBW infants are scanty. CC in infants (<1 year old) has different clinical and pathological features than the pediatric group (1–18 years old). Infantile CC (IFCC) typically appears as sizable cystic masses caused by obstruction in the distal bile ducts. Clinical manifestations include symptoms like cyst enlargement, obstructive jaundice, and in rarer cases, spontaneous rupture, occurring in less than 2% of cases [4]. Several incidences of jaundice, vomiting, acholic stools, and hepatomegaly occur in neonates and infants with CC [5]. The etiology of IFCC is probably associated with bile duct obstruction that causes high proximal bile duct pressure and dilatation initially in the extra-hepatic segment and subsequently in the intrahepatic component [2].
Internal drainage procedures without resection, such as choledochocystoduodenostomy or choledochocystojejunostomy have been previously used and are often associated with high morbidity rates and the need for subsequent surgeries. These methods have been reported to leave the cyst in place, thereby allowing the influx of pancreatic enzymes into the cyst via the anomalous pancreaticobiliary junction. This can lead to calculi formation, anastomotic strictures, and cyst-associated carcinoma [3]. Complete cyst excision with a Roux-en-Y hepaticodochojejunostomy is a safe and feasible surgical procedure for treating CC in infants with massive cystic lesions [5]. However, the optimal timing for the intervention in neonates is debatable. CC is generally treated by performing early resection within the first 3 months of life to prevent cholangitis, spontaneous perforation, progressive cholestasis, liver fibrosis, and malignancy [3]. The incidence rate of liver fibrosis or cirrhosis is higher in IFCC than in CC in children. Early surgical intervention has exhibited satisfactory outcomes in symptomatic IFCC cases. Nevertheless, not all infants diagnosed with CC have prominent symptoms. In a clinically and biochemically asymptomatic patient, surgery might be postponed to decrease the complication rate. A large cohort study reported that the operation might be delayed safely until the age of 6 months or a weight of 6 kg is attained in asymptomatic patients to decrease complication rates and ensure anesthesia safety [6].
Furthermore, for premature or low-birth-weight infants with high surgical risks, the definitive surgery for CC could be postponed by performing initial external biliary drainage followed by delayed primary excision [4]. External drainage delays further progression to liver fibrosis by relieving the obstruction in biliary outflow while awaiting definitive repair. It has been suggested that external biliary drainage should be performed when the patient is a neonate with subsequent delayed primary cyst excision after 1–2 months or in cases of spontaneous perforation with bile peritonitis, severe cholangitis, or a poor clinical condition where performing primary excision is difficult [5]. External drainage can be approached through two methods: image-guided or laparoscopic procedures. When patients exhibit a large cyst, abnormal liver function, coagulopathy, or insufficient response to conservative treatments, image-guided percutaneous external drainage is considered a viable alternative [7]. In our VLBW patient, primary excision was not feasible, so percutaneous external biliary drainage was performed to delay the progression to liver fibrosis and the possibility of spontaneous rupture. IFCC managed using external biliary drainage before definitive treatment controlled early cholangitis and easy cyst dissection at the time of definitive repair. Further, it delays progression to liver fibrosis by relieving biliary outflow obstruction while waiting for definitive treatment [2].
External biliary drainage by depleting bile acid can lead to bile acid deficiency and cause electrolyte imbalance, fat malabsorption, and diarrhea [8]. Therefore, reinfusion of bile through oral refeeding can be used to replenish innate bile acids and restore intestinal barrier function. Bile replacement during external biliary drainage can be beneficial in adults [9]. Further research of bile replacement is needed in infants, especially in VLBWs.
To the best of our knowledge, this is the first to demonstrate feasible management of a large-sized IFCC in VLBW infant. The outcome was favorable. To wait until the patient’s weight increased to an appropriate level to make surgery feasible, we performed external biliary drainage with oral bile refeeding followed by definite cyst excision with Roux-en-Y hepaticoenterostomy.
Conclusion
IFCC must be treated as soon as possible, but it is difficult in VLBW infants due to their small size. Delayed definitive repair surgery with external biliary drainage could be a feasible alternative for managing CC in low-birth-weight infants.
Footnotes
Acknowledgments
Thank you to Dr. Cheng-Wei Chen for her review of the manuscript. Thank you also to Hsinchu Municipal MacKay Children’s Hospital for their contribution.