
Abstract
BACKGROUND:
Congenital Diaphragmatic Hernia (CDH) is a severe congenital anomaly with significant morbidity and mortality. It can be isolated or can be associated with other congenital anomalies, including broncho-pulmonary sequestration (BPS). The association of CDH with BPS (CDH+BPS) is uncommon but has been previously reported, and it can complicate the course of the disease in patients with CDH. We report two cases of CDH+BPS that were recently treated at our CDH-Qatar (CDH-Q) program.
METHODS:
We reviewed CDH-Q program registry to search for CDH+BPS and extracted the data for the identified cases. We also reviewed the previously published literature available on PubMed for similar cases.
RESULTS:
Out of 53 cases of CDH referred to CDH-Q from January 2018 to December 2022, two cases of CDH+BPS were identified, with an estimated prevalence of 3.8% of this association in our CDH population. Both cases were born at term. Case 1 was diagnosed with CDH+BPS postnatally, while case 2 was diagnosed with CDH antenatally but BPS was diagnosed after birth. Both cases underwent a surgical repair of the CDH with resection of the associated BPS, and the histopathology of the resected lung tissue confirmed the presence of BPS in both. Both cases survived to discharge.
CONCLUSION:
The association of CDH+BPS is uncommon; however, it can have significant consequences on the management and the prognosis of patients with CDH. Reporting these cases is important to provide a better understanding of this association and its impact on CDH patients.
Abbreviations
Congenital Diaphragmatic Hernia
broncho-pulmonary sequestration
CDH-Qatar program at Sidra medicine
CDH with associated BPS
neonatal intensive care unit
Chest X-ray
Ultrasound
high frequency oscillatory ventilation
inhaled nitric oxide
day of life
pulmonary hypertension
patent ductus arteriosus
CDH study group
magnetic resonance imaging
Observed to expected total lung volume
heart rate
beats per minute
computed tomography
CDH associated pulmonary hypertension
extracorporeal life support
congenital pulmonary airway malformation
Introduction
Congenital Diaphragmatic Hernia (CDH) is a severe congenital anomaly with significant morbidity and mortality [1–3]. It occurs most commonly on the left side, whereas bilateral hernias are rare and mostly fatal [2]. Reported prevalence varies in different geographical locations, but generally ranges between 1 to 4 per 10000 births [1, 4]. CDH can be an isolated anomaly, or it can be associated with other congenital anomalies in more than 30% of cases [3, 4]. These anomalies include brain malformations, congenital heart disease, broncho-pulmonary sequestration (BPS) and other lung malformations, gastrointestinal anomalies, renal anomalies, and musculoskeletal anomalies [3, 4]. CDH can also be associated with chromosomal and genetic abnormalities in more than 10% of cases [3, 4]. Reported survival of patients with CDH has improved over the last 20 years to more than 70% [3, 5]. The survival is higher for infants with an isolated CDH, while those infants with CDH and associated congenital anomalies tend to have increased morbidity and mortality [4].
In this article, we report two cases of CDH that were treated at CDH-Qatar (CDH-Q) program and were found to have associated BPS (CDH+BPS). CDH-Q program was established in 2018 at Sidra medicine in the state of Qatar, and it receives prenatal and postnatal referrals for all the cases of CDH in Qatar. Over a period of 5 years, from January 2018 to December 2022, 53 cases of CDH were referred to CDH-Q. Unique features of CDH-Q program are the multinational patient population and the higher incidence of associated congenital anomalies (up to 50%) and genetic abnormalities (up to 30%), compared to the reported literature.
Cases
Case 1
A full-term female infant was born at a referring hospital by a vaginal delivery with associated maternal chorioamnionitis. The mother was diagnosed with diet-controlled gestational diabetes and polyhydramnios during pregnancy, however neither CDH nor BPS were suspected prenatally. The infant was born in a poor condition with Apgar scores of 3 and 4 at 1 and 5 minutes, respectively. She was intubated and mechanically ventilated at birth.
Following initial resuscitation, the infant was admitted to the neonatal intensive care unit (NICU) at the referring hospital. She remained hypoxic with signs of poor perfusion on admission. Chest X-ray (CXR) and abdominal ultrasound (US) on admission revealed the diagnosis of a right sided CDH, with herniation of the liver and the small bowel into the right hemithorax, resulting in a significant mediastinal shift to the left side (Fig. 1). A replogle tube was inserted to decompress the distended intra-thoracic bowel. The infant required stabilization of her cardiorespiratory status with high frequency oscillatory ventilation (HFOV), inhaled nitric oxide (iNO), epinephrine, dopamine, and milrinone. She was transferred to Sidra medicine NICU on day of life (DOL) 3.
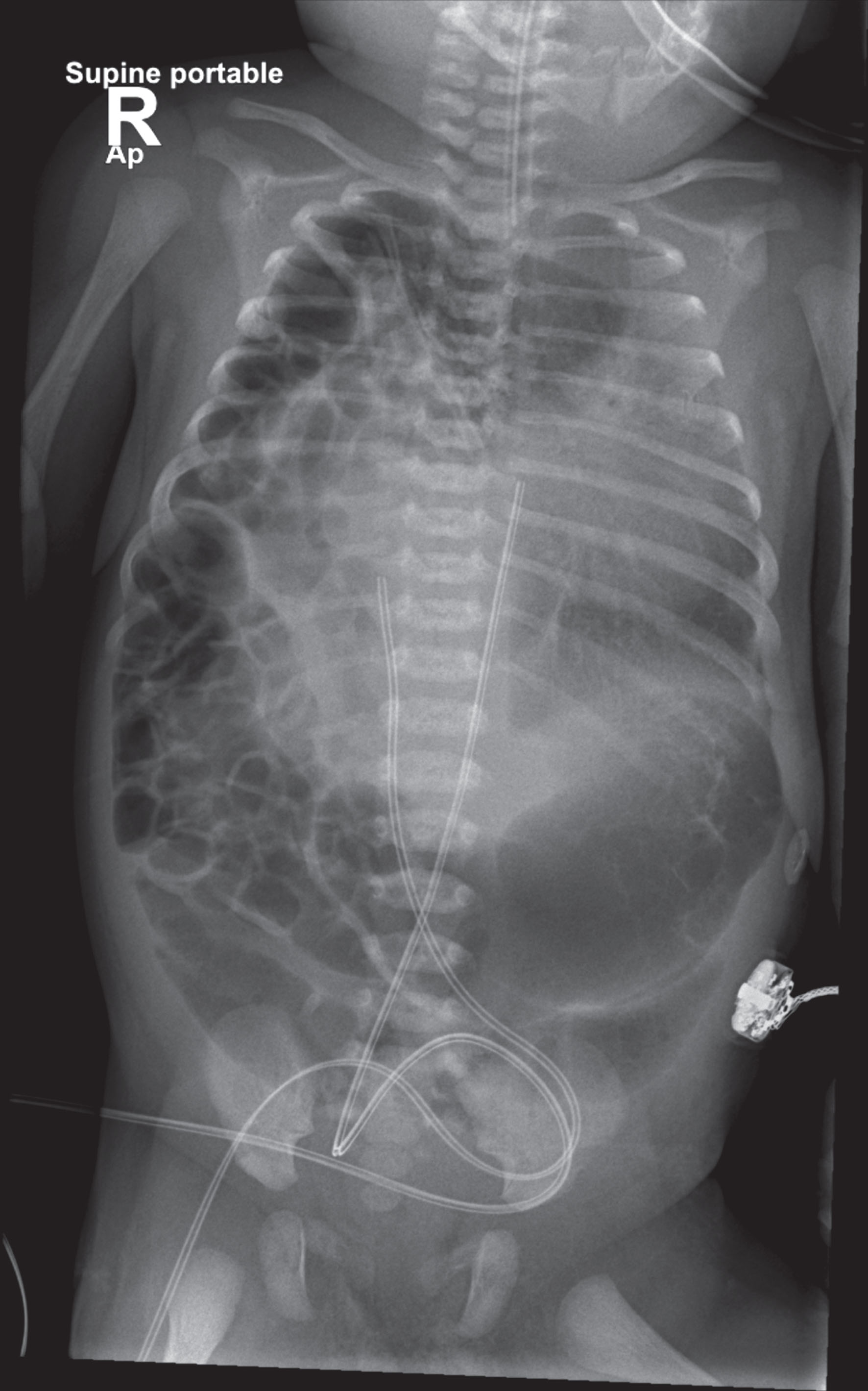
Case 1 –Early postnatal CXR revealing the presence of right sided CDH, with herniation of the liver and small bowel into the right hemithorax, and significant mediastinal shift to the left.
An echocardiogram was done on admission to Sidra NICU, which showed a structurally normal heart with severe pulmonary hypertension (PH), a small patent ductus arteriosus (PDA) shunting right to left, and a moderately dilated right ventricle with depressed function. Alprostadil was started. The clinical condition of the infant gradually improved over the following days, and ventilation and oxygen were weaned gradually. iNO and vasopressors were successfully weaned off by DOL 10. She underwent surgical repair of the CDH on DOL 15.
The infant underwent an open repair of the right CDH via a right subcostal incision. Intra-operatively, she was found to have a large right diaphragmatic defect (Type C based on the CDH study group (CDHSG) classification) [6], with a good anterior rim but an absent posterior rim on the medial side, with herniation of most of the liver, the small intestines, and the large intestines into the thoracic cavity. The infant was also found to have a BPS in the right lung. The abdominal organs were pulled down to the abdomen, the BPS was resected, and the diaphragmatic defect was closed with a patch. The infant tolerated the procedure without any intraoperative or immediate postoperative complications.
Post-operatively, the ventilation was gradually weaned until she was successfully extubated on DOL 25. Enteral feeds were started on DOL 20 and gradually advanced to full feeds by DOL 31. The infant was discharged home on DOL 43.
The histopathological examination of the resected lung tissue showed the appearance of a relatively immature lung tissue consistent with BPS; however, this tissue was supplied by an anomalous pulmonary - not systemic - artery. Of note, postnatal screening studies, including a chromosomal microarray, confirmed the absence of any other congenital or chromosomal anomalies.
A full-term female infant was prenatally diagnosed with a left sided CDH at 23 weeks of gestation. A fetal magnetic resonance imaging (MRI) at 34 weeks of gestation confirmed the presence of a left sided CDH, with a complete absence of the left hemidiaphragm and herniation of part of the liver, the spleen, the stomach, the small bowel, and a significant part of the large bowel. The observed to expected total lung volume (O/E TLV) ratio was 0.47. No other congenital malformations, including BPS, were seen on the MRI.
The infant was born following a planned induced vaginal delivery. At birth, she was noted to be pale and floppy with an irregular breathing and a heart rate (HR) of more than 60 but less than 100 beats per minute (BPM). She was immediately intubated at 2 minutes of life. Following intubation and mechanical ventilation, her color, saturation, and HR improved, and she was transferred to the NICU at 15 minutes of life.
A CXR on admission confirmed the presence of a left sided CDH, with the bowel seen in the left hemithorax and a mediastinal shift to the right side (Fig. 2). The infant required moderate settings of conventional mechanical ventilation after admission, and an echocardiogram showed a structurally normal heart with a moderate pulmonary hypertension and a normal biventricular cardiac function. Initially, the infant needed iNO for pulmonary hypertension but remained otherwise stable. On DOL 3, however, she suffered from worsening of the pulmonary hypertension, a right ventricular dysfunction, and systemic hypotension. The echocardiogram at that time also showed a closing tiny PDA with bidirectional shunting, so Alprostadil was started. iNO was maximized and she was started on milrinone and epinephrine. On DOL 4, considering the continued critical clinical condition, she underwent a surgical repair of the CDH through a left subcostal incision.
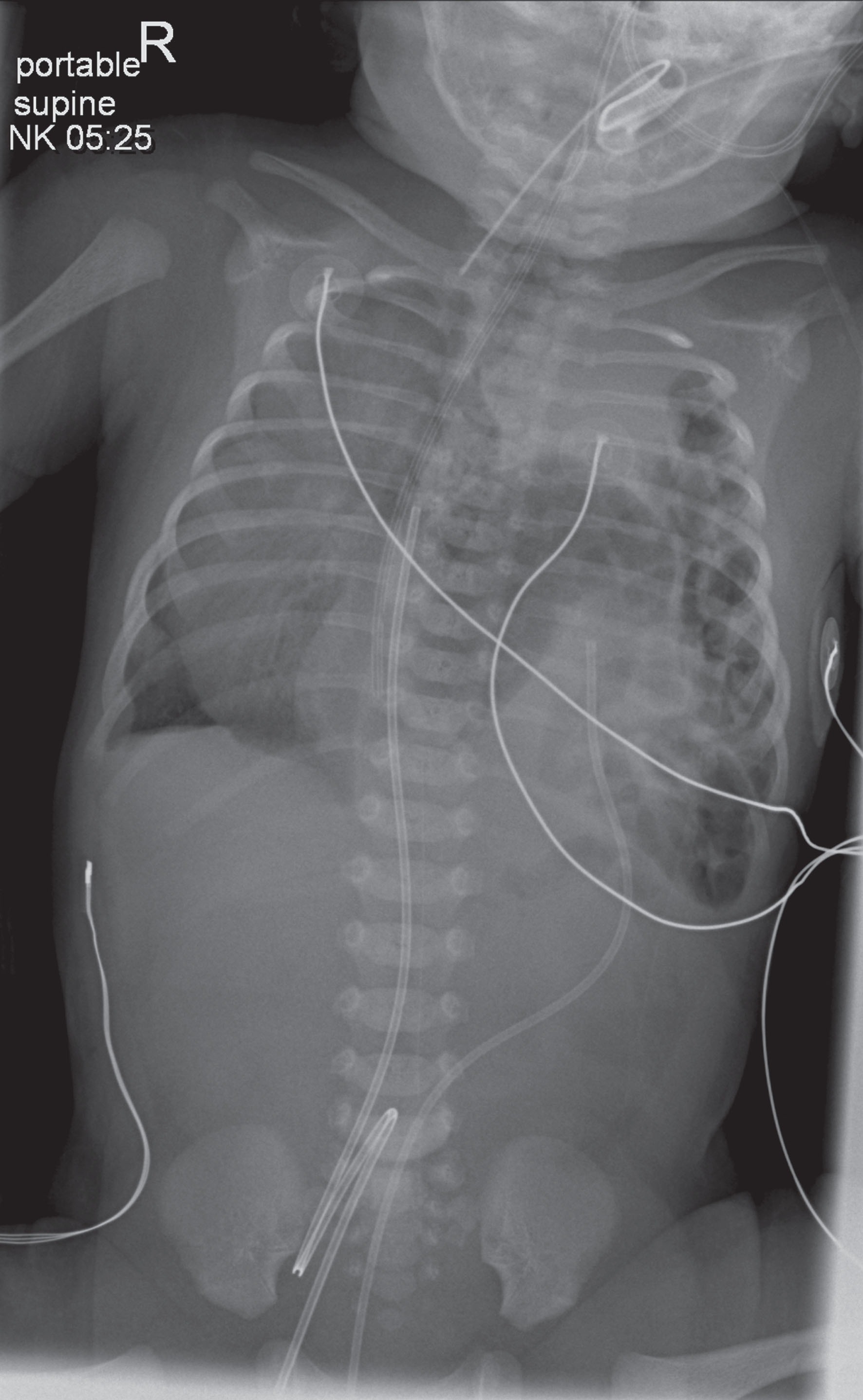
Case 2 –Early postnatal CXR showing the presence of a left CDH with bowel seen in the left hemithorax and mediastinal shift to the right.
Intra-operatively, the infant was found to have a large diaphragmatic defect of type C on the CDHSG classification [6], with herniation of the small bowel, the large bowel, the stomach, and the left lobe of the liver into the left thoracic cavity. She was also found to have a BPS in the lower lobe of the left lung, supplied by a systemic artery coming from the anterior chest wall. The herniated abdominal organs were pulled into the abdomen, the BPS was resected, and the diaphragmatic defect was closed with a patch. The infant tolerated the procedure without any intraoperative or immediate postoperative complications.
As the clinical status and the oxygen requirement improved, iNO was gradually weaned off in the post-operative period. Serial echocardiograms, however, continued to show evidence of pulmonary hypertension, so she was started on intravenous then oral Sildenafil and was discharged home on it on DOL 70. This was later discontinued in the out-patient setting as the pulmonary hypertension resolved.
Due to multiple failed attempts of extubation, the infant underwent an airway evaluation with a bronchoscopy on DOL 26, which showed a mild mid-tracheal narrowing down to the carina with a mild malacia of the bronchus intermedius. She was successfully extubated into non-invasive ventilation on the same day. A chest computed tomography (CT) scan was done on DOL 34 for follow up as the infant was still requiring non-invasive ventilation, and it showed the presence of a moderate sized left pleural effusion, but no obvious compression or narrowing of the trachea or the main bronchi. The non-invasive ventilation was slowly weaned in the following days, till she was completely weaned off any respiratory support by DOL 46. Enteral feeds were started on DOL 9 and gradually advanced to full feeds by DOL 20.
The histopathological examination of the resected lung tissue confirmed the diagnosis of a bronchopulmonary sequestration with a systemic arterial blood supply. Of note, postnatal screening studies, including a chromosomal microarray, confirmed the absence of any other congenital or chromosomal anomalies.
Overview of CDH management
CDH is a complex congenital anomaly that can be challenging to treat. Evidence showed that the management of infants with CDH in a high-volume center and following a standardized evidence-based guideline result in improved outcomes [7]. CDH-Q program was established in 2018 and receives all the cases of CDH in the state of Qatar. Clinical practice guidelines were established with the initiation of the program. These guidelines are based on the international CDH management guidelines published by the CDH EURO consortium [7] and The Canadian Congenital Diaphragmatic Hernia Collaborative [8]. The key recommendations in the CDH-Q guidelines are: Early intubation at birth and avoidance of mask positive pressure ventilation; insertion of an orogastric or a nasogastric tube at delivery to decompress the bowel; gentle ventilation (starting with conventional ventilation then HFOV if indicated) with avoidance of high pressures; permissive hypercapnia; maintenance of an adequate oxygenation (target preductal saturation of 85% to 95%) with avoidance of hyperoxia; and providing adequate sedation while avoiding muscle paralysis unless indicated. In CDH-Q, we also monitor the hemodynamic status of the patient closely using clinical parameters (capillary refill, blood pressure, urine output, and lactate levels) and functional echocardiography, and this monitoring guides the hemodynamic management with fluids, vasopressors, and inotropes as indicated.
The use of iNO in the management of CDH associated pulmonary hypertension (CDH+PH) remains a subject of ongoing debate [2, 7–10]. Data from recent animal and human studies showed both increased and decreased sensitivity of pulmonary arteries to iNO in CDH+PH [8–13]. Similarly, it is also suggested that CDH+PH has a reversible component, resulting from either an imbalance of sympathetic and parasympathetic innervation of pulmonary vasculature or from an imbalance of vasodilator and vasoconstrictor mediators [2, 15]. This reversible component can justify the use of iNO as a first line, with continuation of therapy only if the patient show signs of improvement [2, 13]. Despite its wide use [2, 12], the evidence in the literature that iNO reduces the need for extracorporeal life support (ECLS) or improves survival in patients with CDH is conflicting. Some studies show a possible benefit while others suggest no benefit or even a potential harm [2, 16]. The ongoing clinical trials on the use of iNO in CDH, such as the CoDiNOS (The Congenital Diaphragmatic hernia Nitric Oxide versus Sildenafil) trial [17] and the NoNO (Inhaled Nitric Oxide for Congenital Diaphragmatic Hernia) trial [18], may offer clarity on the subject in future.
In CDH-Q clinical practice guidelines, the first line treatment of CDH+PH in the absence of left ventricular dysfunction is iNO, starting at a dose of 20 parts per million. If the patient responds to iNO, the treatment is continued and iNO is later weaned gradually based on the clinical and echocardiographic signs of improvement. If the patient does not respond, iNO is stopped. The second line therapies for CDH+PH in CDH-Q guidelines are milrinone (if there is evidence of cardiac dysfunction) and Sildenafil. These guidelines are also consistent with the CDH management guidelines published by the CDH EURO consortium [7] and The Canadian Congenital Diaphragmatic Hernia Collaborative [8].
The optimal timing of surgery is another controversial aspect of CDH management [2, 8]. CDH-Q guidelines recommend that the repair is done electively once medical stabilization is achieved, as evidenced by a normal systemic blood pressure, a sub-systemic pulmonary pressure, a fraction of inspired oxygen (FiO2) of less than 50% to maintain the target preductal saturation, and normal lactate levels and urine output. Failure to meet these criteria within 2 weeks is an indication to attempt repair or to redirect care to palliation. Surgery can also be performed while on ECLS. These recommendations are consistent with the CDH management guidelines published by the CDH EURO consortium [7] and The Canadian Congenital Diaphragmatic Hernia Collaborative [8].
CDH with associated BPS (CDH+BPS)
Congenital lung anomalies are generally uncommon, with a reported incidence of up to 1 in 2500 livebirths [19]. Broncho-pulmonary sequestration (BPS) is the second most common congenital lung anomaly, after congenital pulmonary airway malformation (CPAM), accounting for 0.15% to 6.4% of all cases [20]. Histopathologically, BPS is comprised of an immature, non-functioning lung tissue that, unlike in CPAM, does not communicate with the tracheobronchial tree [19]. BPS is also characterized by its systemic blood supply [19, 20], however it can rarely be supplied by an anomalous artery arising from the pulmonary arterial circulation [21]. Based on the location of the BPS compared to the normal lung tissue and the presence of a distinct pleural covering, BPS is divided into two different types: Intra-lobar and Extra-lobar [22]. An intra-lobar BPS is located adjacent to the normal lung tissue and is not separated from it by a pleural covering. An extra-lobar BPS, on the other hand, has a distinct pleural covering that makes it anatomically completely separated from the normal lung tissue [20, 22]. While both types are histologically similar [19], extra-lobar BPS is less common but is more commonly associated with other congenital anomalies in more than 60% of patients, including CDH, cardiac anomalies, pulmonary hypoplasia, and gastrointestinal anomalies [22, 23].
While some of the cases of prenatally diagnosed BPS regress before delivery [24], and many can be asymptomatic at the time of birth [25], BPS can sometimes be associated with short term and long term complications. BPS can impair lung growth and development prenatally, resulting in respiratory distress or failure at birth due to pulmonary hypoplasia [24, 26]. BPS might also require surgical resection in the neonatal period, resulting in loss of lung tissue [24]. In addition, BPS can cause intra-pulmonary left to right cardiac shunting, resulting in cyanotic episodes and high output heart failure [24, 27]. Lung hypoplasia and intrapulmonary shunting are particularly important when the BPS is associated with CDH, as they can complicate the effect of CDH on lung development and heart function [26]. In the long term, unresected BPS can be complicated by infection, possible risk of malignancy, and spontaneous pneumothorax [24].
The association of BPS with CDH is uncommon but has been previously reported in the literature [23, 28–39]. The incidence of CDH+BPS was estimated to be 3.4% in a recent study by Coughlin et al. [28]. On the other hand, CDH is reported to be the most common congenital anomaly associated with extra-lobar BPS, occurring in 16–40% of cases [22, 23]. This association between CDH and BPS can be explained by the theory that that the development of BPS, typically at 4–5 weeks of gestation, mechanically interferes with the fusion of the diaphragm and pleuroperitoneal folds closure, resulting in CDH [23].
In this article, we report two cases of CDH+BPS from the CDH-Q program. With 53 patients treated in CDH-Q over 5 years, the estimated incidence of CDH+BPS in CDH-Q is around 3.8% of patients with CDH. This is similar to the incidence reported in the study by Coughlin et al. [28]. In this study, CDH+BPS association was reported to be more commonly seen in patients with larger defects (type C and D on the CDHSG classification) [6] compared to smaller defects [28]. The findings in our cases are consistent with this conclusion, as both infants had a type C defect.
The study by Coughlin et al. also found CDH+BPS to be associated with more need of ECLS and higher mortality [28]. Despite these findings, other studies suggested a potential survival advantage in patients with CDH+BPS [30, 34]. While this is difficult to prove with the small number of reported cases, a proposed pathophysiological mechanism for this potential protective effect is that the presence of BPS prevents the herniation of a significant portion of the abdominal content into the thoracic cavity [23, 34], allowing for a less abnormal lung development. Both of our cases support this theory as both survived to discharge with good outcomes. In addition to that, case 2 had an O/E TLV of 0.47 on fetal MRI, which is above the reported median for survivors in the literature and correlates with a lesser degree of pulmonary hypoplasia and a better prognosis postnatally.
While the prenatal diagnosis of CDH+BPS has been reported, many of these cases are not diagnosed till the postnatal period [28]. The inability to diagnose CDH prenatally in some cases can be potentially explained by the theory that BPS acts as an anatomical barrier during pregnancy, preventing the herniation of the abdominal content, thus obscuring the diaphragmatic defect [23, 34]. This was the case in our first infant, in whom the diagnosis of both CDH and BPS was only made postnatally. Prenatal diagnosis of BPS itself has been also reported in the literature [40], but it can be difficult to differentiate BPS from other thoracic space-occupying lesions, including CDH [40]. In our second case, CDH was diagnosed prenatally but the BPS was not seen. In this case, we hypothesize that the BPS did not act as an anatomical barrier and the abdominal content herniated into the thoracic cavity, which then obscured the BPS and made it difficult to be seen on both prenatal US and fetal MRI. This demonstrates that the presence of BPS and CDH together makes the prenatal diagnosis of either of them more challenging. More research is needed to explore this effect to improve the prenatal diagnosis of both entities, as this will improve the prenatal counseling of families and help to define the true incidence of CDH+BPS [28].
Some of the previous reports of CDH+BPS reported the presence of other congenital anomalies as well. These included major cardiac anomalies [28, 38], CPAM [29, 39], laryngo-tracheo-esophageal cleft [39], intestinal malrotation [26], gastric volvulus [35], Renal anomalies [23], and chromosomal abnormalities [28]. Taking into consideration the fact that CDH itself is commonly associated with other anomalies [4], it is unclear if the presence of BPS in association with CDH results in a higher chance of having other anomalies or not. Both of our cases did not have any other associated congenital or chromosomal anomalies. The study by Coughlin et al. did not find any significant difference in the rate of association of CDH with either major cardiac anomalies or chromosomal abnormalities when a BPS is present [28]. Nevertheless, anticipating and identifying the presence of other anomalies in addition to CDH+BPS is very important as it can significantly affect the prognosis and help improve prenatal family counseling.
Conclusion
CDH+BPS is a rare occurrence; however, it can significantly affect the management and prognosis of infants with CDH. Proper identification of these two anomalies, ideally prenatally, and improved management of this association are of utmost importance. Reporting of the prenatal and postnatal course, the management, and the outcome of such cases may help to further define this complex association.
Disclosure statement
The authors have no disclosures.