
Abstract
Renal tumors (i.e., abnormal growths within the kidney) encompass both benign and malignant entities. The pathologist plays a key role in determining the nature of the growth (i.e., benign or malignant) by evaluating the tumor with a microscope. It is essential to distinguish between these two categories, as malignant tumors possess the potential to spread or metastasize throughout the body, while benign tumors typically follow a less aggressive course and stay limited to the organ. Benign growths may be followed clinically or surgically removed. Excision is often considered if the benign mass, by virtue of its location, is pressing on the bowel or a blood vessel. The removal of a benign growth via surgical excision is typically curative. Therefore, the need for accurate differentiation between benign and malignant renal tumors cannot be emphasized enough as it is imperative for appropriate patient care and management. All this information is recorded and reported in the pathology report. The report is shared with the surgeon, or the primary care physician, who will review and discuss the findings with the patient. In this review we will discuss the histomorphology and challenges in diagnosis of some of the more frequently occurring benign renal neoplasms.
INTRODUCTION
Renal masses are frequently, although not exclusively, detected incidentally with imaging studies. The radiologists’ reports often summarize their suspicions about the renal mass being benign or malignant. Occasionally the radiologic findings are equivocal and in those cases the mass is reported as suspicious with a need for tissue biopsy or excision for definitive diagnosis. Alternatively, if the mass is small without a concern for malignancy, the recommendation may be “watchful waiting” where the patient remains in surveillance and imaging is performed at regular intervals (i.e., every 6 to 12 months) to watch the progress/growth rate of the mass. If the mass is growing at a slow rate, then they suspect it to be a “low-grade” lesion that could potentially be benign. If the growth is rapid, then the surgeon may recommend excision suspecting it to be a malignant lesion. Ultimately, with all this information, the patient will need to make the choice on how to proceed.
When a partial or total nephrectomy is performed, where a part of the kidney or the entire kidney is excised, respectively, the pathologist assesses the physical appearance and unique characteristics of the mass, as well as the extent of its involvement. The differentiation between benign and malignant renal neoplasms can be challenging in a subset of cases due to morphologic overlap but is critical as it can have a profound impact not only on the patient’s psyche, but also on the clinical management and prognosis. The World Health Organization (WHO) classification of renal neoplasms includes well established benign and malignant entities; it also mentions “emerging entities” which have been recognized by pathologists but require additional investigation regarding pathologic features, molecular alterations and clinical outcomes. Table 1 lists accepted and emerging benign renal tumors in the WHO, many of which (mostly the epithelial tumors) will be discussed in this review. Accordingly, we will delve into entities that share histological and immunohistochemical features but exhibit distinct variations that set them apart from malignant tumors.
Commonly Diagnosed Benign Renal Neoplasms.
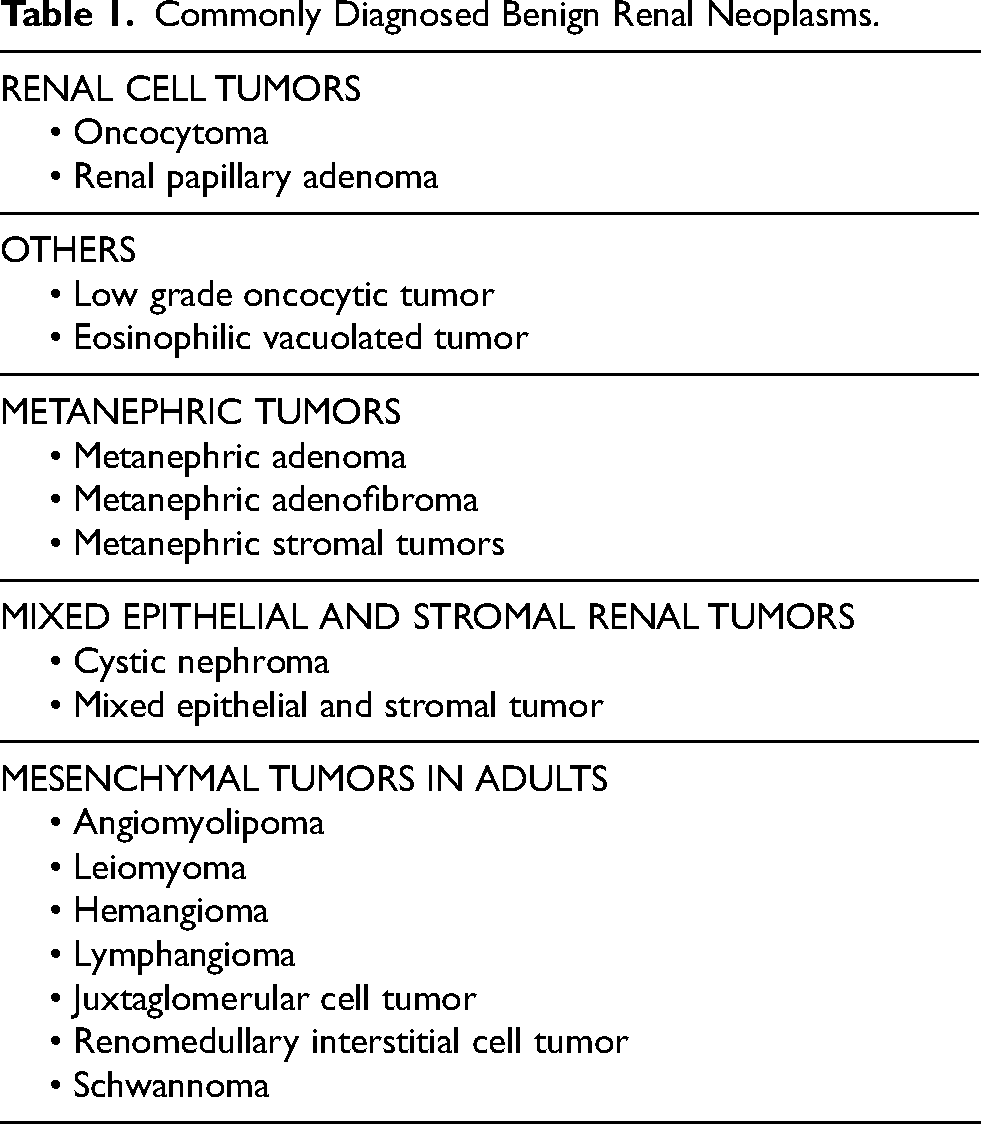
RENAL ONCOCYTOMA
Renal oncocytoma (RO) is a benign neoplasm and accounts for approximately 6–10% of renal tumors, affecting a broad age range with a mean age of 62 years and a male-to-female ratio of 2 : 1 [1–6]. Although they frequently occur sporadically, rare hereditary cases and associations with Birt–Hogg–Dubé (BHD) syndrome have been reported. Multiple oncocytomas can also occur simultaneously, a condition referred to as “renal oncocytosis” [7]. Oncocytomas typically arise from the outer portion of the kidney (i.e., the renal cortex), and can occasionally extend into adjacent tissues (i.e., adipose tissue) with no change in overall behavior.
Oncocytoma, on gross inspection, is typically a well-circumscribed unencapsulated tumor that is mahogany-brown or yellow-tan in color with or without a central stellate or peripheral scar [3–6]. While oncocytomas have a favorable prognosis, they can sometimes be challenging to distinguish from other renal tumors. When reviewed under the microscope, it has a solid, nested, or microcystic architecture composed of pink or eosinophilic cells rich in mitochondria (Fig. 1) [5, 6]. Oncocytomas exhibit relatively distinctive features, including dense eosinophilic cytoplasm, round nuclei, and small prominent nucleoli. However, these microscopic features are not entirely specific and may be seen in other benign and malignant renal tumors, such as low-grade oncocytic renal tumors (LOT) and chromophobe renal cell carcinoma (ChRCC), respectively [8–12]. The distinction of these tumor entities can be confounding in a smaller subset of cases, especially on renal core biopsy specimens, which are typically small and may not be representative of the entire lesion. A special staining technique (i.e., immunohistochemistry) can be used for challenging cases with oncocytomas staining diffusely positive for KIT/CD117 and scattered rare cells positive for cytokeratin (KRT7); this is distinct from LOTs and ChRCCs, which are KIT negative and KRT7 diffusely positive and KIT and KRT7 diffusely positive, respectively [13–15].

Renal oncocytoma.
Emerging Benign Renal Epithelial Neoplasms: Low grade oncocytic tumor (LOT) and Eosinophilic vacuolated tumor (EVT)
As briefly discussed above, many renal tumors can show overlapping histologic features with oncocytoma and ChRCC. Such ambiguous tumors were previously given several names like “oncocytic RCC, not otherwise specified (NOS)”, “oncocytic, low-grade RCC”, “renal oncocytic tumor, NOS”, “unclassified oncocytic tumor”, “oncocytic tumor with uncertain/low malignant potential”, and “borderline tumor” (or with “borderline features”) [8–10]. Despite the presence of low grade cytology, it was not always clear if these cases were benign or malignant. However, with recent technological advancements, these ‘ambiguous’ tumors have been analyzed further with molecular techniques, and ultimately resulted in the discovery of two distinct benign tumor entities: LOT [13, 14] and eosinophilic vacuolated tumor (EVT) [13, 18]. It is likely that LOTs and EVTs were previously (mis)diagnosed as RO, ChRCC or other eosinophilic/oncocytic RCCs. Surveillance and/or surgical excision is often the preferred treatment approach for oncocytoma, LOT and EVT, with a favorable prognosis, as these tumors tend to be low-grade and have a low risk of recurrence or metastasis. Long-term follow-up monitoring would only be needed to ensure early detection of any potential recurrence or complications.
LOW GRADE ONCOCYTIC TUMOR (LOT)
LOT is a relatively uncommon, eosinophilic renal tumor that may be more often seen in young individuals. They mostly occur sporadically or very rarely in a hereditary setting associated with tuberous sclerosis (TSC). The histopathologic features of LOT have been described as an entity with overlapping features of renal oncocytoma and ChRCC. LOT is typically a small, solid tumor with mahogany-brown to tan cut surface. It exhibits solid and compact nested growth; focal tubular and/or trabecular growth may be present [13–15]. The eosinophilic tumor cells have round to ovoid nuclei and although the nuclear membranes lack significant irregularities, they may show perinuclear clearing/halos like those seen in ChRCC (Fig. 2). The histomorphology reveals sharply delineated, edematous areas with scattered, irregularly arranged cells referred to as “boats in a bay” [13, 14]. These tumors lack prominent nucleoli and other high-grade features, such as necrosis and increased mitotic activity.

As mentioned, LOT is diffusely positive for KRT7 and is typically negative for KIT/CD117; some studies have demonstrated positive GATA3 expression in LOTs [16]. On electron microscopy, a high-resolution microscope that allows pathologists to identify the contents of the tumor cell cytoplasm but not often used as a contemporary diagnostic tool, LOTs exhibits abundant, closely packed cytoplasmic mitochondria, like oncocytoma [3, 4, 11]. Molecular studies reveal TSC/MTOR pathway gene mutations in LOT; this is distinct from oncocytoma. Distinguishing LOT from oncocytomas is clinically insignificant as both are benign neoplasm; however, the association of MTOR pathway mutations in the former may be important in a small subset of patients [16–18]. The distinction of LOT from ChRCC/other RCCs is important to avoid mislabeling a malignant diagnosis for a benign neoplasm specifically because on imaging it shows up as an avid and typically heterogeneously enhancing solid renal mass which may suggest an RCC [20].
EOSINOPHILIC VACUOLATED TUMOR (EVT)
EVT is a relatively rare renal neoplasm that tends to occur more frequently in adults, with a median age typically ranging from 40 to 60 years. Although typically solitary and sporadic, they can rarely be seen in patients with TSC syndrome [21]. EVTs are solid tumors that typically lack a cystic component and are without a well-formed capsule. Histologically, tumor cells are characterized by abundant eosinophilic cytoplasm and distinctive intracytoplasmic vacuoles. The tumor cells typically have round nuclei with large prominent nucleoli (Fig. 3); some cells may demonstrate multinucleation. Immunohistochemical staining can help differentiate EVT from other renal tumors especially LOT, oncocytoma, and ChRCC. The immunoprofile of EVT includes KIT/CD117 diffuse positivity and KRT7 expression in scattered tumor cells; the distinction from oncocytoma (which demonstrated a similar KIT/KRT7 expression pattern) includes immunoreactivity for Cathepsin K and CD10 in EVT but not typically in oncocytoma. EVTs, unlike oncocytoma, but similar to LOTs, have been shown to harbor MTOR pathway mutations [21].
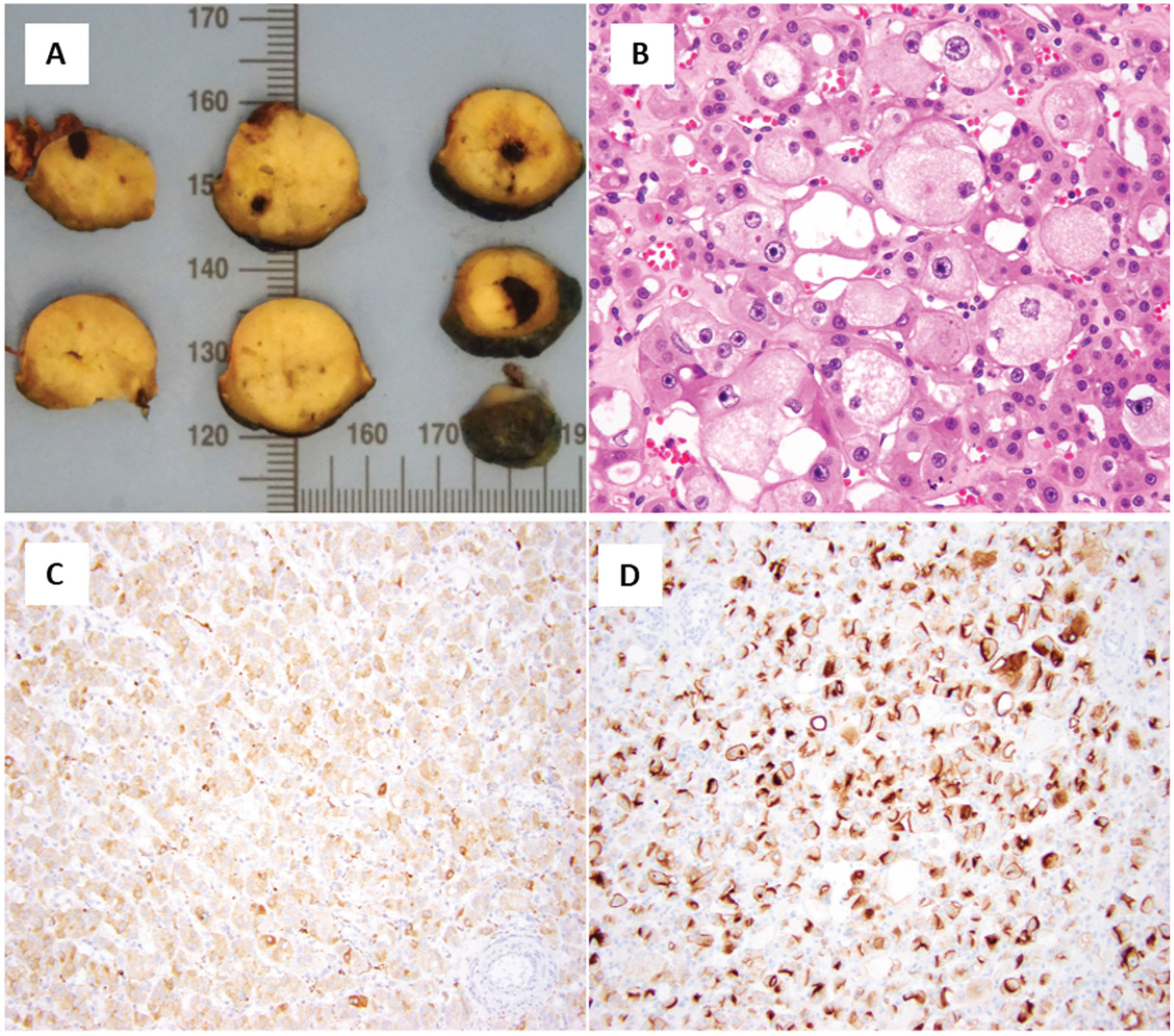
Occasionally, it is still difficult to completely distinguish non-descript low grade renal oncocytic tumors in the kidney due to overlapping morphologic and immunophenotypic findings. In these situations, the recommendation by the genitourinary pathology society (GUPS) is to use “oncocytic renal neoplasm of low malignant potential, NOS.” GUPS also recommends that the term “hybrid oncocytic tumor” be reserved only for renal tumors associated with familial mutations; such tumors are typically bilateral and multifocal as seen in syndromic (i.e., hereditary) settings [15].
RENAL PAPILLARY ADENOMA
Renal papillary adenomas (PAs) are primarily observed in elderly patients, particularly those aged 70–90 years, and are also found in 10% of individuals aged 21–40 years [1, 2, 24]. They are more common in people with certain kidney conditions like renal vascular disease, glomerulosclerosis, acquired cystic disease and those undergoing kidney transplantation for end stage kidney disease [25–27]. PAs typically arise in the renal cortex (i.e., the outer rim of the kidney, beneath the renal capsule). They can be solitary, or multiple. PAs are ill-defined lesions that lack a capsule and blend into the adjacent kidney tissue. By definition, PAs are typically≤15 mm in diameter [1, 2]. As they are small, they are not always distinctly visible on imaging studies or on gross inspection and are frequently found in kidney resections performed for other causes (tumors, end stage kidney disease or at autopsy). When viewed under the microscope, PAs are of a low histological grade and are characterized by pale cytoplasm, round to oval nuclei, monotonous nuclei with minimal variation in size, and lack mitoses. In keeping with their name, PAs demonstrate papillary, tubular, or tubulo-papillary growth patterns (Fig. 4).

The most important entity this subset of tumors can be mistaken for is papillary renal cell carcinoma, which are usually > 15 mm and are encapsulated. It has been suggested that PAs could be the precursor lesions to PRCC and may possibly represent a continuum of the same biological process, especially the PAs arising in the background of papillary RCC. Nevertheless, their definitive relationship is not completely understood, and more research is being performed to consolidate this theory of evolution [28, 29].
Papillary RCC, which will be further discussed in the accompanying review on renal tumors with papillary architecture in this special edition, but in brief can have an overlapping histologic appearance with PA, especially in low-grade areas. In contrast to PA, papillary RCC may present as a distinct large mass with extensive hemorrhage, variable areas of necrosis and cystic degeneration. Histologically, tumor cells can vary in size and tumor cell may appear more basophilic (i.e., blue) or eosinophilic (i.e, pink).
Immunohistochemical stains for KRT7, CD10, and AMACR will not help distinguish PA from papillary RCC, as these three biomarkers are typically diffusely positive in both neoplasms [30]. Given the indolent nature of PA with virtually no risk of spreading, monitoring and surveillance are typically advised to evaluate for any changes or concerns [31]. Accordingly, PAs often do not require surgical intervention, and the presence of one or more in a donor kidney does not exclude renal transplantation [27].
METANEPHRIC ADENOMA
Metanephric adenoma is another benign renal tumor that is commonly encountered in the fifth or sixth decade of life with a significant predominance in females [32]. Clinical presentations may include incidental discovery, abdominal pain, or hematuria. Interestingly, a small subset of patients can display erythropoietin-induced polycythemia which essentially means there is an overproduction of red blood cells due to the influence of erythropoietin, which can create vascular complications because of the increased density of blood [32, 33]. On gross inspection tumors are typically solid, well-circumscribed but unencapsulated. They can attain considerable size and therefore mimic an RCC. Histologically, metanephric adenomas contain small round blue cells with round nuclei, surrounded by minimal cytoplasm that are reminiscent of embryonic type cells called primitive metanephric tubular epithelium. Architecturally, they form closely packed acini/tubules or angulated ducts, and some demonstrate long branching and abortive glomeruli-like structures. Frequently, calcified bodies, called dystrophic or psammomatous calcifications form when calcium gets deposited on damaged or injured tissues (Fig. 4).
Immunohistochemistry assists in diagnosis, with positive staining for WT1 and CD57, and frequently a lack of KRT7 [33]. Metanephric adenomas often harbor BRAF mutations; accordingly, expression of BRAF by immunohistochemistry supports the diagnosis [34, 35]. Distinguishing metanephric adenoma from a histologically similar appearing tumor called ‘solid low-grade papillary RCC’ is mainly by immunohistochemistry as the latter stains KRT7 positive and typically BRAF negative [36, 37]. The other entity in the differential diagnosis is the epithelial-predominant nephroblastoma (i.e., Wilms tumor) which can be distinguished by younger age presentation, presence of pseudocapsule, high-grade nuclear features and lack of the BRAF mutation in most cases [38]. Although metanephric adenomas are generally benign, rare coexistence with nephroblastoma or papillary RCC has been reported [39].
MIXED EPITHELIAL AND STROMAL RENAL TUMORS
This family of tumors consists of two main entities. One is the mixed epithelial and stromal tumors (MEST), and the other is adult cystic nephroma (CN). Some believe they are on the same spectrum as both are benign and share some similar histologic findings [1, 2, 40–44]. These unilateral renal tumor masses may arise in the medulla, and are typically asymptomatic, but when symptoms occur, they often manifest as hematuria and abdominal pain. In some cases of large MEST or CN, the lesions may grow at the cost of normal renal function.
MESTs (Fig. 5) are frequently seen in middle aged to perimenopausal women at a median age of about 50 years [45–47]; history of chronic hormonal treatment has been suggested as a potential trigger, despite rare cases reported in male patients [46, 50, 51]. They are typically solitary, well circumscribed tumors, with variable solid and cystic components. The tumors are biphasic in nature and include epithelial cells that line cystic spaces that may resemble thyroid follicle-like spaces and stromal cells that comprise the fibrous tissue intervening between the cystic spaces. The stroma can resemble ovarian stroma which is immunoreactive for estrogen and progesterone receptors [40–42]. Radiologically, because of the complex cystic nature of these lesions, MESTs (70% have solid enhancing components) more than cystic nephromas are frequently classified as Bosnaik III cysts which indicates they are indeterminate cystic masses that may suggest malignancy [48]. Although a majority of these tumors are benign, rare reports of malignant transformation have been reported, including sarcomatous differentiation, MEST with rhabdoid features, undifferentiated components, and carcinosarcoma [49–54].
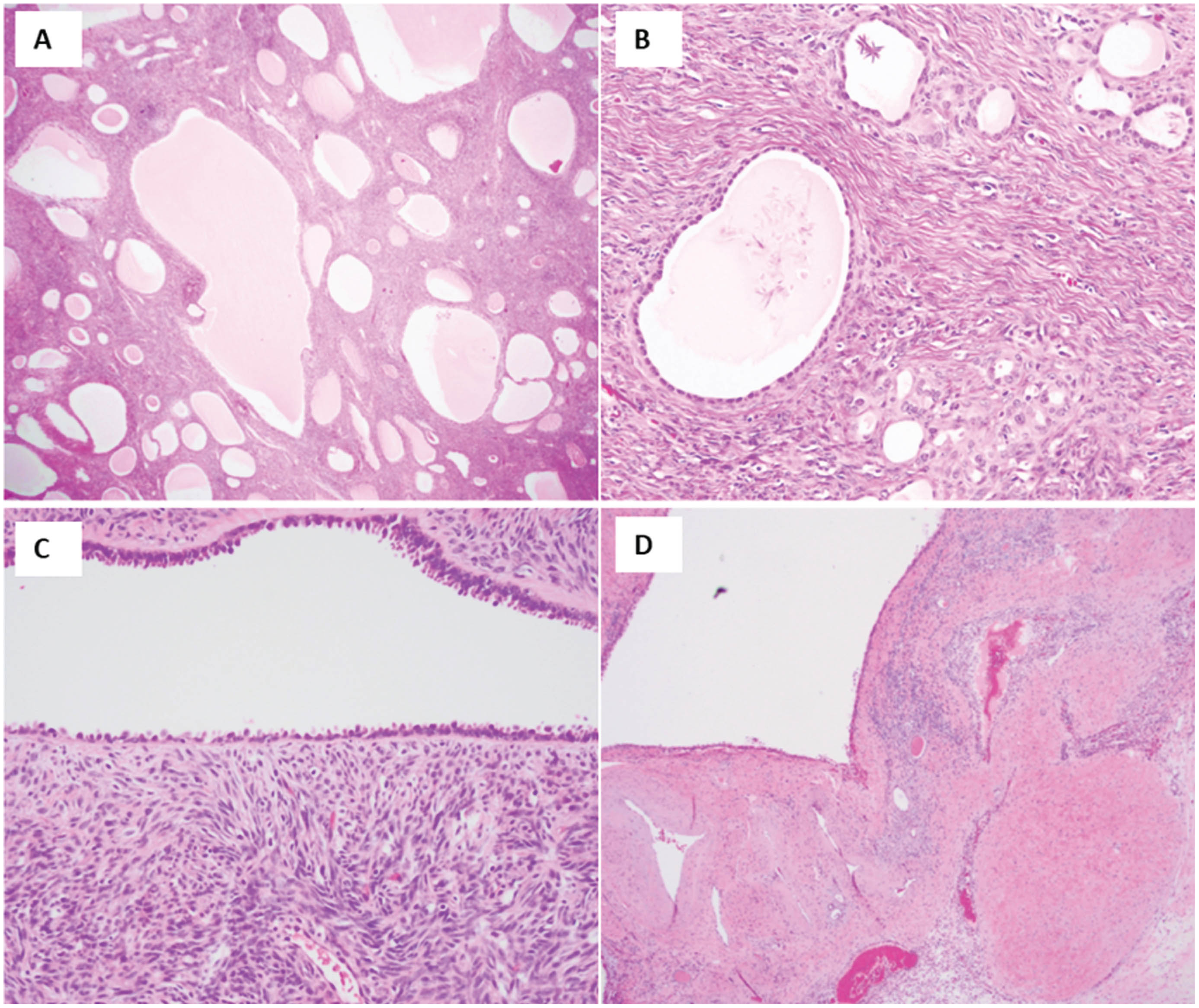
Mixed epithelial and stromal tumor.
DICER1 tumors and synovial sarcoma are considered in the differential diagnosis of mixed mesenchymal epithelial stromal tumors due to their overlapping histopathological characteristics, including a spindle cell stroma/neoplasm which can create diagnostic challenges. Mutations in the DICER1 gene are implicated in a range of pediatric and adult tumors [55–61], including certain ovarian and renal neoplasms. When a renal mass exhibits a biphasic pattern with spindle cell stroma and glandular epithelial elements, a DICER1 tumor should always be considered as there are genetic implications. In contrast, synovial sarcoma is a mesenchymal malignancy composed of spindle cells and occasionally epithelial components that can rarely occur in the kidney [62–64]. Moreover, synovial sarcoma often expresses epithelial markers such as cytokeratins, further complicating the differentiation from MEST. However, the presence of the SYT-SSX fusion gene in synovial sarcoma, which results from the characteristic t(X;18) translocation, provides a critical diagnostic clue and can be identified through immunohistochemistry and/or molecular testing, distinguishing it from MEST and DICER1-associated tumors.
Cystic nephroma (Fig. 6) are comprised of non-communicating epithelium-lined cysts without a major solid/stromal component which as previously mentioned show estrogen and/or progesterone receptor positivity in the majority of cases [65–67]. In cystic nephromas, the cells lining the fluid-filled cysts are often flat, cuboidal or hobnailed (i.e., protrude in the cystic spaces). As mentioned, both tumors are exceedingly rare and generally benign, but malignant transformation, usually of the spindle cell stromal component may occur and has been reported in rare cases of MEST, leading to an aggressive clinical course. Therefore, resection may be necessary to confirm that it is indeed benign which is critical for clinical management of the patient [40–42, 66].

Cystic nephroma.
CONCLUSION
In conclusion, the range of benign renal epithelial tumors represents a diverse and complex spectrum of renal neoplasms that requires careful evaluation. The morphologic overlap between benign and malignant renal masses can be challenging; however, careful examination plus ancillary studies such as immunohistochemical can significantly aid the pathologist in making an accurate diagnosis. Understanding the growth patterns, cytologic similarities and differences, and molecular alterations of these tumors is critical for optimal patient management, as the distinction between a benign and malignant mass can significantly impact treatment decisions and prognosis. Therefore, recognizing the unique characteristics of benign renal epithelium plays a key role in guiding clinical strategies, providing reassurance to patients, and ensuring appropriate therapeutic interventions.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments.
FUNDING
The authors report no funding.
AUTHOR CONTRIBUTIONS
Drs. Kandukuri and Karne have equally contributed to this manuscript.
CONFLICTS OF INTEREST
Drs. Kandukuri and Karne have no conflicts of interest to report.