
Abstract
Renal cell carcinoma (RCC) has long been found to be responsive to immunotherapy. While high dose interleukin-2 resulted in some durable remissions, this treatment has largely been replaced by immune checkpoint inhibitor therapy, due to the safer toxicity profile and emerging evidence for long term remissions. However, the majority of patients continue to face disease progression and death from metastatic RCC. Chimeric antigen receptor T-cells (CAR T) represent the next step in immunotherapy for this malignancy and hold promise for a higher rate of durable remissions. The realization of this therapeutic strategy for RCC will require identification of the best tumor antigen and T cell modifications and will depend on achieving remissions with an acceptable toxicity profile. This review summarizes current CAR T-cell treatment targets and clinical trials for metastatic RCC, highlighting the potential therapeutic impact as well as obstacles to successful development.
INTRODUCTION
Renal cell carcinoma affects more than 75,000 men and women in the United States each year, leading to approximately 13,000 deaths in 2020 [1]. Initially this was recognized as an immune responsive tumor, and the mainstay of treatment was high-dose interleukin-2 and interferon gamma (Fig. 1). While the former was associated with durable remissions with a median duration of 24 months there was also a high toxicity rate sometimes necessitating treatment in the intensive care unit due to capillary leak syndrome which at times led to death [2] (Fig. 1). Because of the specialized treatment requirements, interferon was more widely used and was the comparator arm in the landmark trials of early targeted therapies against vascular endothelial growth factor (VEGF) such as sunitinib [3] (Fig. 1). The VEGF inhibitors became the dominant treatment until the immune checkpoint inhibitors (ICI) were found to have strong efficacy in RCC (Fig. 1). One of the first ICIs that was found to be effective in metastatic RCC was nivolumab, which targets the programmed death 1 (PD-1) checkpoint [4]. This agent was found to be more effective than everolimus in the second and third-line setting, inducing objective responses in approximately 25% of patients with nearly a third of these responses lasting at least 12 months [4]. The combination of PD-1 inhibition with cytotoxic T-lymphocyte associated antigen 4 (CTLA-4) inhibition achieved higher response rates and was found to have superior overall survival (OS) compared to sunitinib in first-line therapy for metastatic RCC [5]. Notably patients whose RCC features placed them in the International Metastatic Renal Cell Carcinoma Database Consortium (IMDC) good risk stratum benefitted more from sunitinib, though overall ipilimumab plus nivolumab was associated with longer OS. Still, a majority of patients did not experience objective response with the ICI combination. More recently, combined therapy of a VEGF TKI and ICIs such as axitinib/pembrolizumab, cabozantinib/nivolumab, and lenvatinib/pembrolizumab have emerged as additional FDA approved first line treatment options for this disease (Fig. 1) [6–9].

Evolution of renal cell carcinoma treatments.
Despite these great advances in mRCC treatment, many patients who progress after treatment with ICIs and/or TKIs have no other treatment options with established clinical benefit and are in need of other treatment modalities. Recently, the development of chimeric antigen receptor (CAR) T-cells has emerged as a novel immunotherapy with strong clinical benefit in hematologic malignancies [10–12]. As a result, there has been much excitement to see if this treatment can also be applied to solid tumors including kidney cancers such as renal cell carcinoma. In this manuscript we will discuss 1) basic principles of CAR T-cell treatments in current clinical application, 2) potential challenges in utilizing these treatments for RCC, 3) ongoing and future directions for RCC CAR T-cell clinical trials.
BASIC PRINCIPLES OF CHIMERIC ANTIGEN RECEPTOR T CELLS
CAR T cells: Structural components and function
CAR T-cell treatments are a type of adoptive cell therapy currently used to treat hematologic malignancies [13]. This is done by collecting the patient’s own T-cells and then genetically reprogramming them in vitro to express CARs which are synthetic tumor-specific receptors that will ideally recognize and kill the antigen-expressing tumor cell [12–14]. CAR T-cells are synthetic modular proteins consisting of four components, 1) an extracellular target binding domain which can recognize and bind the tumor cell specific antigen, 2) a hinge domain, 3) this is then linked to a transmembrane region that anchors the CARs to the cell membrane, 4) an intracellular signaling domain that activates the T cell on antigen binding and its cytotoxic activity (Fig. 2) [12]. The current FDA approved CAR T-cells that are being used in the clinic are individually manufactured after leukapheresis from a cancer patient, typically using viral vectors to alter the T-cell receptor, followed by expansion ex vivo and selection to achieve a subset of cells would target cancer cells and have robust cytotoxicity against them [12]. After undergoing a rigorous quality control validation process these newly manufactured personalized CAR T-cells are then infused back into the patient with the goal of promoting a self-sustaining anti-cancer response. Due to the complex manufacturing process, it can take up to two weeks before the patient can receive their CAR T-cell infusion [12].

Structural schematic of basic CAR T-cell (Created with Biorender®).
Because T-cells form a major component of the anti-tumor immune effector cell population, engineering them to identify and be activated by the presence of cancer has the prospect of becoming a potent therapy. For example, T-cells genetically modified to target CD19 have created high rates of durable remissions in patients with chemotherapy refractory acute lymphoblastic leukemia [14]. This clinical benefit has led to the US FDA and European Medicine Association (EMA) approvals for the CD19 targeting CAR T-cell therapies: tisagenlecleucel (KYMRIAH®) for pediatric and adult B-cell ALL and adult relapsed/refractory diffuse large B-cell lymphoma (R/R DLBCL), and axicabtagene ciloleucel (YESCARTA®) for R/R DLBCL, transformed follicular lymphoma, and primary mediastinal large B-cell lymphoma [10, 14]. These encouraging results have created enthusiasm for studying CAR T-cell therapy in a broader range of solid tumors, including renal cell carcinoma.
The first major challenge of developing CAR T-cell therapy in solid tumors has been identifying optimal tumor antigens that are ideally strongly but selectively expressed on tumor cells with minimal or no expression on normal host tissues. In hematologic malignancies, CD19 is an optimal target as B cell elimination is essentially the only “on target, off-tumor” toxicity against normal host tissues which is fairly well tolerated. In contrast, most solid tumor antigens have some level of expression on normal tissues leading to on target, off tumor toxicities. Another challenge has been tumor antigen heterogeneity in solid tumors which can lead to decreased efficacy in the treatment of solid tumors [15]. Nevertheless, multiple CAR T-cell therapies are undergoing early clinical testing for solid tumors as this method holds much promise.
A detailed discussion on potential barriers to effective CAR T-cell therapy is beyond the scope of this manuscript. For those interested in this topic please refer to the excellent review by Rafiq et al. [16]. We will focus briefly on its main limitations that would need to be overcome before being widely used in the treatment of mRCC or any solid tumor.
LIMITATIONS OF CAR T-CELL THERAPY
The general challenges of implementing widespread use of CAR T-cell therapy can be categorized into complex manufacturing and high financial costs and toxicities.
Complex manufacturing and high financial costs
As discussed above, current FDA approved CAR T-cells require a complex individualized manufacturing process where the cancer patient undergoes leukopheresis for T-cell collection. This is then followed by shipment of the collected patient’s T-cells to a central facility for genetic modification into the appropriate cellular product that is then expanded ex vivo and finally shipped back for infusion. This is a very complex process that can take up to two weeks under optimal circumstances. Unfortunately, this process is vulnerable to logistical mishaps (ie. delays in shipments and contamination during cell manufacturing) delaying delivery of the final product. For example, in a phase 1-2a clinical trial looking at the safety and efficacy of tisagenlecleucel for relapsed B-cell ALL, a total of 92 patients were screened and enrolled but only 75 (81.5%) patients were able to undergo the CAR T-cell infusion [14]. A total of 17 patients (18.5%) were unable to get the final CAR T-cell product due to manufacturing issues (n = 7), dying while waiting (n = 7), or adverse effects (n = 3) [14].
In addition, to the complex manufacturing process that has to be customized for each patient there is also a high financial cost for this treatment. The estimated wholesale acquisition cost for tisagenlecleucel (KYMRIAH®) or axicabtagene ciloleucel (YESCARTA®) range from approximately $400,000 to $500,000 [17, 18]. However, these estimates do not include, costs associated with the inpatient hospitalization for the required pre-treatment conditioning chemotherapy, CAR T-cell infusion services, and possible post treatment hospitalizations for adverse effects. A recent study done by Prime Therapeutics attempted to better determine the total cost of CAR T-cell treatments by analyzing the integrated medical and pharmacy claims data on 74 patients who had received tisagenlecleucel (KYMRIAH®) or axicabtagene ciloleucel (YESCARTA®) starting at 30 days prior to CAR T-cell administration through 56 days after the cellular infusion [19]. What they found was that the total cost of care including the more extended period was much higher with an average of $700,000 with as many as 12% of these patients exceeding $1,000,000 [19]. Furthermore, it was also noted that 39% of the patients progressed on these treatments [19]. These numbers suggest that the estimated per patient total cost for CAR T-cell treatments may be much higher in the $700,000 to $1,000,000 range [19]. The extremely high financial cost of this therapy in relation to its limited clinical benefit is a significant barrier that will need to be overcome before it can become a widely utilized treatment [20].
CAR T cell toxicities
The toxicities related to CAR T-cell therapy are adverse effects of the anti-tumor T-cell response which include cytokine release syndrome (CRS), immune effector cell associated neurologic toxicity (ICANS) and less commonly hemophagocytic lymphohistiocytosis/macrophage activated syndrome (HLH-MAS) [12]. There is also the potential for immune compromise due to the lymphodepletion chemotherapy that is typically administered prior to CAR T-cell infusion [12].
CRS occurs in 40–50% of patients undergoing CD19 directed CAR T-cell treatment [10, 14], and can manifest immediately following CAR T-cell infusion, which is why CAR T-cell treatment is often administered in the inpatient setting. However, it should also be noted that the peak incidence for CRS is between 2–7 days [21]. The predominant symptoms of CRS are fevers, chills, hypotension, and hypoxia and in severe cases has led to multiorgan failure [21]. Once it was recognized that CRS symptoms are largely related to elevated levels of IL-6, the use of anti-IL-6 antibodies such as tociluzumab and siltuximab were found to very successfully mitigate the syndrome, preventing severe cases and restoring organ function [21]. Although clinical data is limited, the reduction in IL-6 levels has not been observed to be associated with a significantly reduced anti-tumor effect of the CAR T-cell treatment [21, 22]. Immune effector cell-associated neurotoxicity syndrome (ICANS), is associated with the severity of CRS but manifests later, often 2-3 weeks after CAR T-cell infusion [23–25]. This can occur in approximately 60% of patients and include headache, lethargy, confusion, aphasia, or even seizures [23–26]. Primary management is with dexamethasone as IL-6 inhibitors are often ineffective [23, 24]. Finally, in rare cases approximately 1% of patients have been found to experience HLH-MAS in some phase I studies with CAR T-cell treatments in myeloma [27] and prostate cancer [28]. This is a very serious hyperinflammatory syndrome characterized by CRS, hemophagocytosis, renal and liver failure which if untreated can at times be lethal [29].
In solid tumor trials, toxicities related to “on-target, off-tumor” effects of CAR T-cells have been observed. For example, one of the first CAR T-cell targets in mRCC was the transmembrane protein carboxy-anhydrase-IX (CA IX), which exhibits nearly ubiquitous, high level expression in clear cell RCC tissue samples [30]. However, it is also expressed at low levels in some normal tissues including the bile duct. Due to expression in the normal bile duct, patients enrolled in the phase I clinical trials undergoing CA IX CAR T-cell treatments were found to experience frequent high-grade liver toxicities limiting its use [30]. Attempting to mitigate the toxicity by administering CA IX antibodies prior to CAR T-cell infusion did reduce the expected hepatotoxicity, but unfortunately also eliminated a clinically significant anti-tumor response [31]. The limitations of on target, off tumor toxicity demonstrated by this case has shown that the challenges in identifying the appropriate specific tumor associated antigen. Additionally, conditional inhibition or activation of the CAR is being evaluated in fourth generation CAR T-cells reduce toxicity against normal tissues [32].
While immune checkpoint inhibitors can cause immune related adverse events, which can be severe or even lethal, the management has been well defined and the risk-benefit profile has been favorable enough for widespread use across all oncology settings. This has led to a change in practice where high dose interleukin 2 is now seldomly used due to its much more difficult toxicity profile and required inpatient hospitalization and specialized management. As a result, the eventual use and the frequency and severity of CAR T toxicities in RCC will also be compared against the immune checkpoint inhibitors.
USING CAR T CELLS IN RCC: CHALLENGES IN DESIGN AND IMPLEMENTATION
Early experience with RCC targeting CAR T cell therapy
As discussed above despite having higher enrichment in malignant RCC tissues, targeting CA IX led to significant on-target, off-tumor toxicities affecting the hepatobiliary system leading to early termination of the clinical trial [30, 31]. Another target that has been attempted is vascular endothelial growth factor (VEGF) Receptor 2. Many RCC tumors have an alteration in the Von Hippel Lindau gene, which results in overexpression of VEGF and cancer addiction to signaling through this pathway, is nearly ubiquitous in RCC [33]. The high rate of response to small molecule inhibitors targeting this pathway provided further rationale for targeting this antigen using CAR T-cell therapy, with the hope of eliciting more durable remissions. Unfortunately, the VEGFR2 CAR T-cell clinical trial (NCT01218867) was terminated when treated patients were found to have no objective treatment response [34].
The reasons for lack of efficacy of early attempts at CAR T-cell therapy for RCC have not been fully defined. However, we can extrapolate from studies of CAR T-cell therapy in hematologic malignancies and other solid tumors on the possible mechanisms. One key mechanism which has been identified is the loss/downregulation of tumor antigen which is also known as “antigen escape” [15]. In patients with refractory ALL that were treated with CD19 targeting CAR T-cells 70–90% of the patients had an initial durable response [35, 36]. However, later in in these studies approximately 30–70% of the patients were observed to have developed recurrent disease coinciding with the decrease/loss of CD19 or antigen escape [35, 36]. Antigen escape from CAR T-cell treatments has also been observed in other malignancies such as multiple myeloma and glioblastoma [37–40].
Another potential mechanism for lack of efficacy is the immunosuppressive tumor microenvironment (TME). In solid tumors the TME often contain lower number of endogenous tumor-infiltrating T-cells compared to lymphoid tissues housing hematologic malignancies [41, 42]. In addition, solid tumors tend to reside in an immune suppressive microenvironment containing regulatory T (Treg) cells, myeloid-derived suppressor cells (MDSCs), and immune suppressive ligands such as PD-L1 [41]. Several studies have shown that tumors from RCC patients tend to have an immunosuppressive TME containing abundant Treg cells and MDSCs [43–46]. One can extrapolate that this immunosuppressive TME could also contribute to inhibition of CAR T-cell activity in RCC.
Finally, there are physical and stromal barriers that may also play a role in lack of CAR T-cell treatment efficacy [15]. One such physical barrier is heparin sulfate proteoglycan (HSPG) in order to pass into the tumor [15, 47]. In order to overcome this hurdle to cell trafficking CAR T-cells that were engineered to express heparinase could degrade the HSPG barrier and promote tumor infiltration and subsequent antitumor killing [48]. Another barrier is that many solid tumors have a hypoxic TME that may adversely impact the viability of effector T-cells and immune cells [49, 50]. One proposed mechanism is that hypoxia promotes ATP breakdown which then leads to extracellular accumulation of adenosine in the tumor microenvironment [50]. The excess adenosine has been found to target A2AR and A2BR receptors expressed on T cells surface which can then interfere with TCR signaling and thus inhibit the antitumor activity of T cells [50].
RENAL CELL CARCINOMA: ONGOING AND FUTURE TRIALS
Despite the above mentioned challenges and setbacks there are several ongoing efforts to utilize CAR T-cells for the treatment of mRCC (Table 1). Amongst these efforts CD70 appears to be a promising target for CAR T-cell treatment in RCC patients.
Ongoing active CAR T-cell clinical trials for patients with advanced metastatic renal cell carcinoma
CD70 has been identified as a potential target antigen with high specificity and expression in clear cell RCC while having low expression in normal tissues. This is a membrane protein which binds to the tumor necrosis factor receptor (TNFR) known as CD27. While CD70 functions on immune cells as a costimulatory signal it is also highly expressed on clear cell RCC [51, 52]. Furthermore, CD70 is rapidly internalized on antibody binding which has made it an attractive target for drug targeting in clear cell RCC [53, 54]. Both CD70 and CD27 have been targeted with antibody-drug conjugates. AMG172, which is anti-CD27 IgG1 conjugated to the cytotoxic DM-1 (semi-synthetic maytansine) was found to have limited efficacy in a phase one clinical trial resulting in two patients (5.4%) with a partial response, 6 patients (16.2%) with stable disease, and 13 patients (35.1%) who had progressive disease [55]. Of note there was a single patient who had a 79% reduction in the sum of the target lesions which lasted 12 months [55]. However, the agent was not pursued further as it was found to have dose limiting toxicities including thrombocytopenia, hepatic injury, myocardial infarction, and renal failure [55]. A second antibody-drug conjugate agent has also been tested clinically, SGN-CD70A, which targets CD70 and is linked to pyrolobenzodiazepine [56]. In the phase 1 trial for SGN-CD70A one partial response was seen at the highest dose level [56]. However, this agent also faced toxicity issues with thrombocytopenia and abdominal pain with 28% of subjects discontinuing treatment due to adverse events [56]. Due to the limited clinical benefit and toxicity of both AMG172 (anti-CD27 antibody drug conjugate) and SGN-CD70A, they have not been further studied in a follow up phase 2 clinical trial.
Despite issues with the antibody drug conjugates targeting CD70, CAR T-cells have been developed to target this cell surface marker in RCC. Preclinical work with CD70-directed CAR T cells showed significant anti-tumor activity in murine models [57]. This has led to two current phase one CD70 directed CAR T-cell clinical trials. Both of these trials use allogeneic CAR T-cells to target CD70 in clear cell RCC. This is significant because the current FDA approved CAR T-cell products are autologous and require patient-specific cell collection which is then followed by a complex manufacturing process which can take weeks to infuse into the patient. Many patients who are eligible for CAR T-cell treatment have advanced heavily pre-treated malignancies that continue to grow leading to a decline in their performance status and unfortunately become ineligible for their cellular infusions. In fact it was found that in one study approximately 30% of patients who had started the treatment plan to receive current FDA approved autologous CAR T-cells were unfortunately not able to get the treatment due to progressive disease [11]. Furthermore, it has also been hypothesized that the autologous CAR T-cells derived from relapsed cancer patients who have undergone multiple lines of therapies might be prone to early immune exhaustion [58]. Therefore, the current two allogeneic CAR T-cell products being tested in RCC attempt to overcome these hurdles by being readily available for use off the shelf and by being derived from healthy donors without any prior history of cancer who do not have early immune exhaustion.
The first is an anti-CD70 allogeneic CRISPR-Cas9-engineered T cell (CTX130) which is being tested in adult patients with advanced (unresectable or metastatic), relapsed or refractory clear cell RCC developed by CRISPR Therapeutics®. CTX130 is currently being tested in an ongoing phase 1 clinical trial (NCT04438083) evaluating for safety, efficacy, pharmacokinetics (PK), pharmacodynamics (PD), and maximal tolerated dose (MTD). The allogeneic CTX130 CAR T-cell is designed to be a product readily available “off-the-shelf” for infusion and avoids the above mentioned time consuming complex manufacturing process. Per information provided by the company there are three modifications that have been employed with the use of CRISPR/Cas9 technology that enhance the efficacy of these CAR T-cells. The first is the use of CRISPR/Cas9 to integrate into the TCR alpha constant (TRAC) locus allowing for precise control of the integration site and copy numbers of the CAR sequence. This contrasts with the traditional use of attenuated lentivirus or adenoviruses to deliver the CAR construct, which can lead to variability in copy numbers of the delivered CAR sequence and unanticipated site of integration leading to mutagenesis in the resulting T-cells. Although not yet proven, the hope is that these advantages of CRISPR/Cas9 technique will yield greater safety and efficacy. Furthermore, CRISPR/Cas9 technology is used to knock out both the T-cell receptor and MHC I receptor in the CTX130 cellular products (Fig. 3). This decreases the risk of graft vs host disease and donor T-cell rejection, allowing for the allogeneic product.
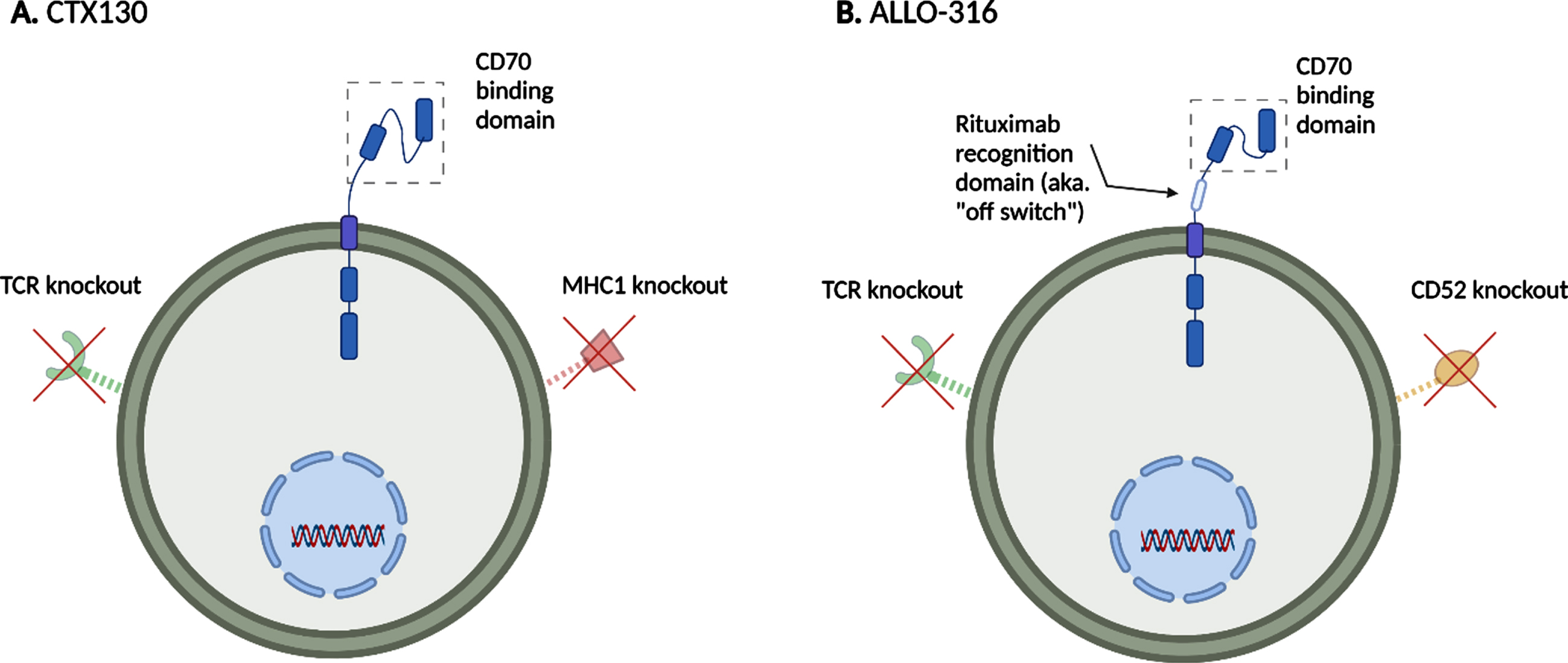
Structural schematic CD70 targeting CAR T-cells for metastatic renal cell carcinoma patients. A) CTX130 allogeneic CAR T-cell. TCR and MHC1 receptors have been knocked out using CRSPR-CAS9. B) ALLO-316 allogenic CAR T-cell. TCR and CD52 receptors have been knocked out using TALENS. (Created with Biorender®).
A second ongoing RCC CAR T-cell trial is the TRAVERSE study (NCT02830724), testing the allogeneic CAR T-cell ALLO-316 which also targets CD70. The TRAVERSE study utilizes the standard fludarabine/cyclophosphamide lymphodepletion intensified with ALLO-647 (anti-CD52 monoclonal antibody) for preconditioning. The proposed mechanism for how ALLO-647 can be beneficial is that by targeting CD52 in the host this will work synergistically with the fludarabine/cyclophosphamide and further deplete the host T-cells which would allow better integration of the donor CAR T-cells [59]. It should be noted that the ALLO-316 CAR T-cells were designed using transcription activator-like effector nucleases (TALENS) [60] to knock out the TRAC region and CD52 [59]. This allows the ALLO-316 CAR T-cell products to be resistant to the CD52 inhibition of ALLO-647 treatment. In addition, the ALLO-316 product has a CAR that is composed of a fully human scFv specific to CD70, a CD19 rituximab-based off switch, and 4-1BB and CD3ζ signaling domains to help potentiate the T-cell response (Fig. 3).
The on-target off-tumor toxicity profile of targeting CD70 has not yet been described. As with all solid tumor CAR T trials, there is potential that repeated dosing or immune adjuncts may be needed to achieve the kinds of durable remissions that are achieved with CAR T-cell therapy in hematologic malignancies. The results of both CD70 CAR T trials are eagerly anticipated and will hopefully bring the field closer to utilizing cellular immunotherapy for RCC.
CONCLUSION
Renal cell carcinoma is a tumor that is susceptible to immunotherapies. In the past decades there has been much progress in improving patient outcomes using various immunotherapy agents such as cytokines, immune checkpoint inhibitors, and combinations VEGF targeted therapies with immune checkpoint inhibitors. However, metastatic RCC still remains an incurable disease, creating a significant public health burden leading to efforts to develop new treatments such as CAR T-cells. Further work will need to be carried out to make CAR T-cells an effective and safe treatment for the treatment of RCC.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments.
AUTHOR CONTRIBUTIONS
Y.L. and T.B.D. wrote and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
FUNDING
Y.L. is supported by the American Cancer Society (Grant #P063266).
CONFLICTS OF INTEREST
Y.L. has served as an advisory board consultant for EMD Serano and Pfizer. T.B.D. has served as an advisory board consultant for Bayer, Janssen Oncology, Seattle Genetics, Abbvie, Exelixis, Advanced Accelerator Applications, and Astra Zeneca. T.B.D. is an Editorial Board Member of this journal, but was not involved in the peer-review process of this paper, nor had access to any information regarding its peer-review.