
Abstract
Increasing evidence suggests a potential role for infectious pathogens in the etiology of synucleinopathies, a group of age-related neurodegenerative disorders including Parkinson’s disease (PD), multiple system atrophy and dementia with Lewy bodies. In this review, we discuss the link between infections and synucleinopathies from a historical perspective, present emerging evidence that supports this link, and address current research challenges with a focus on neuroinflammation. Infectious pathogens can elicit a neuroinflammatory response and modulate genetic risk in PD and related synucleinopathies. The mechanisms of how infections might be linked with synucleinopathies as well as the overlap between the immune cellular pathways affected by virulent pathogens and disease-related genetic risk factors are discussed. Here, an important role for α-synuclein in the immune response against infections is emerging. Critical methodological and knowledge gaps are addressed, and we provide new future perspectives on how to address these gaps. Understanding how infections and neuroinflammation influence synucleinopathies will be essential for the development of early diagnostic tools and novel therapies.
Plain Language Summary
This review explores how infections might contribute to the development of Parkinson’s disease and other synucleinopathies. It highlights evidence that microbial pathogens may trigger neurodegeneration by causing neuroinflammation. We emphasize the complex relationship between infections, genetics, and neurodegeneration, and discuss how understanding these connections could lead to earlier diagnosis and new treatments. In this review we also identify key knowledge gaps, and we suggest areas for future research.
INTRODUCTION
Age-related neurodegenerative diseases such as synucleinopathies are likely caused by a complex interplay between host genetics and the environment. Within this environment, there is a lifelong accumulation of environmental exposure to toxicants and pathogens. The totality of environmental exposure (the exposome), lifestyle and demographics might be crucial determinants in the development of neurodegeneration. 1
Synucleinopathies are a category of age-related progressive neurodegenerative diseases, characterized by the intracellular accumulation of alpha-synuclein (αSyn) containing aggregates. 2 This group includes Parkinson’s disease (PD), dementia with Lewy Bodies (DLB), multiple system atrophy (MSA), and other atypical forms.3,4, 3,4 Historical epidemiological studies have often suggested an infectious etiology of parkinsonism. Now, there is renewed attention for a possible infectious etiology as recent studies show that virulent agents can act as triggers of synucleinopathy, and that genetic risk modulates the pathogenic response to inflammatory or infectious agents. 5
Several bacterial, viral, and fungal pathogens are associated with the onset of sporadic cases of synucleinopathy. 5 However, whether infections causally contribute to the pathogenesis of synucleinopathies or help to exacerbate existing pathology remains to be established. This rapid evolving field of research has recently presented evidence for the involvement of αSyn and other PD risk factors in the innate and adaptive immune response. Specifically, it is shown that the host response against infections is mediated via αSyn. αSyn is a pre-synaptic protein with high expression in the brain, but also functions as a key protein in the response to pathogenic and commensal microorganisms.
Here, we summarize epidemiological and experimental observations linking infections with synucleinopathies. We propose that infections can trigger inflammatory processes that could contribute to αSyn pathological aggregation and associated neurodegeneration. We also explore the possibility that the pathological agent, the infection site, and the host genetic background could affect αSyn protein conformation and function, and as consequence, the neurodegenerative process.
EPIDEMIOLOGICAL EVIDENCE
An infectious etiology of parkinsonism
An infectious etiology for synucleinopathies has been discussed for decades. Early observations of viral epidemics followed by an outbreak of parkinsonism have led to a long-held hypothesis that parkinsonism could be of infectious origin, and it has long been discussed whether viruses could be the initiating etiology of primary PD or if viruses could be causative for secondary parkinsonism.
A notable example and a historic conception of a potential infectious etiology of PD is the postencephalitic parkinsonism pandemic in the years following the Spanish flu. The Spanish flu, caused by a severely virulent H1N1 influenza A virus, affected millions of people worldwide and resulted in a global pandemic. After the outbreak of the Spanish flu, in 1918–1919, many patients subsequently developed encephalitis lethargica, a condition that is characterized by high fever, delayed physical and mental response and lethargy.6,7, 6,7 Although these symptoms from encephalitis lethargica only occurred sometime after infection it was clear that the onset pattern of encephalitis lethargica inherently followed the one of the influenza pandemics.
During this early phase of encephalitis lethargica, other neurological manifestations concurrently developed as patients experienced phases of delirium or episodes of psychotic manifestations. 8 Encephalitis lethargica could almost be seen as a precursor of neurological disease as a bout of encephalitis was almost invariably followed by postencephalitic parkinsonism. 8 Here, the classical signs of parkinsonism would include a mask-face like appearance, paralysis agitans, tremor, rigidity of all the muscles, and shuffling gait. 7 These symptoms could develop in all infected individuals, including young adults. 9 In the following decades, it was thus speculated that a link between viral respiratory infections and neurological diseases such as PD might exist. 10 However, these links became muddled because of a profusion of clinical signs and the long time between infection and the onset of neurological symptoms in which the association between parkinsonism and influenza was simply forgotten.
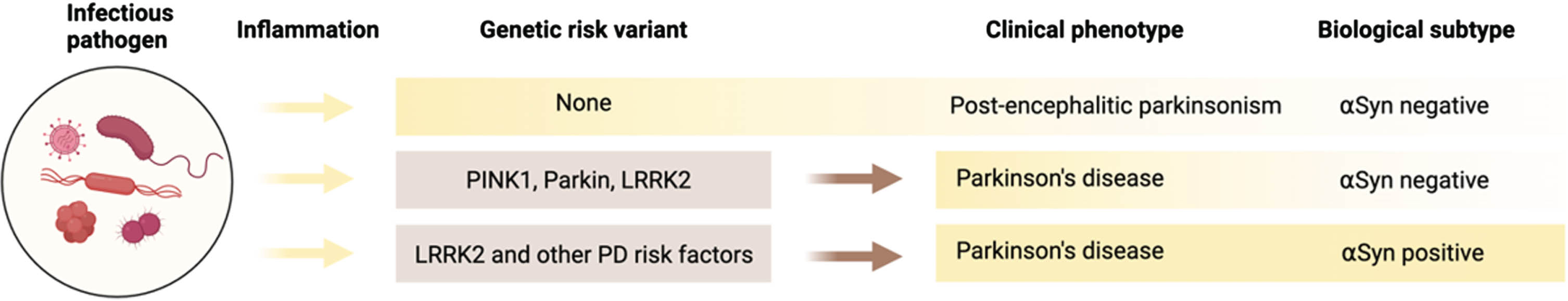
Nevertheless, pathological examinations of patients with postencephalitic parkinsonism, some of which were done more recently,11–14 revealed that PD-related brain regions of patients infected with the Spanish flu were indeed severely affected. In infected cases, a remarkable hyaline degeneration with loss of blood vessels was observed in the striatum and more generally in the basal ganglia,6,15, 6,15 Vascular subacute inflammation was sometimes seen, especially in the brainstem, but in the absence of signs of chronic or active neuroinflammation elsewhere in the brain 16 . From these historic cases, the most notably affected region was the substantia nigra, a region typically affected in synucleinopathies, showing loss of pigmented dopaminergic neurons and gliosis.11,12,17, 11,12,17 Maybe most striking was that this neurodegeneration within the substantia nigra and the basal ganglia was accompanied by the presence of rounded or oval viral inclusions bodies but most often there were numerous cellular neurofibrillary tangles.11–14,18, 11–14,18 Importantly, the presence of αSyn positive inclusions in these cases was investigated, with recent immunohistochemical techniques and αSyn-specific antibodies, but αSyn pathology could not be shown.11–13 In cases of postencephalitic parkinsonism, and in the absence of synucleinopathy, it seems that based on these findings, there is more evidence for a tauopathy-related type of parkinsonism after influenza infection rather than predominantly synucleinopathy-related PD. It also suggests that based on its underlying biological mechanisms, postencephalitic parkinsonism might represent a unique entity in the biological classification of PD (Fig. 1) (we further discuss other implications of infections on the clinical classifications of PD below).
The development and onset of neurological symptoms after influenza infection varied. The onset of parkinsonism after influenza infection ranged from days to a more prolonged onset with cases that would show neurological symptoms not until decades after infection 8 . Symptoms also developed when the infected person was considered to have fully recovered from the infection. The establishment of infection-related encephalitis lethargica or postencephalitic parkinsonism as a clinical entity thus rests on the abundant epidemiological, clinical, and pathological descriptions during that time. Of note, the delayed onset of neurological symptoms is in line with the natural history of PD, where a long prodromal phase precedes the occurrence of the more typical cardinal symptoms.
Along those lines, case reports of infection with other viruses have also been reported to cause postencephalitic dopamine-responsive parkinsonism with alterations in the basal ganglia and neurodegeneration of the substantia nigra.19–21 Several of these case reports include infections with SARS-CoV-2,22–25 enterovirus,26–29 Japanese encephalitis virus,30–32 St. Louis encephalitis, 33 West Nile virus,34–37 HIV,38–40 and dengue viral infections.19,41, 19,41 Importantly, this spectrum of viruses demonstrates that development of neurological symptoms and brain pathology after infection does not require the virus to enter brain tissue or to replicate in neurons as some of these viruses are not neurotropic.
An infectious etiology of idiopathic Parkinson’s disease
Although viral post-encephalitic cases show many clinical similarities with PD, these cases of viral encephalitis with parkinsonism are etiologically distinct from idiopathic PD. The mechanisms leading to selective degeneration of the basal ganglia and the substantia nigra in the absence of overt synucleinopathy are in stark contrast to the natural history of PD and the progression of pathology in idiopathic cases.
Nevertheless, links between various microbial infections as causative or precipitating triggers of idiopathic PD have recently received renewed attention, especially in the wake of recent epidemiological studies. In a systematic and population-based case control study from Denmark, various types of infections were examined for their potential link with PD. An association was shown between the onset of PD and infection with influenza when the infection occurred more than 10 years before clinical diagnosis. 42 Similar findings were observed in a second and independent systematic population-based study. Here, cases from Finland and the UK were studied. 43 A significant association was again found between infection with influenza and the subsequent development of PD if the time between infection and diagnosis was 5 years. 43
Another notable association from these studies is the link between PD and urinary tract infections. 42 Aside from influenza infections, urinary tract infections were found to be the only other type of infection associated with an increased risk of PD more than 10 years after infection. 42 Interestingly, similar findings were found for urinary tract infections and MSA. 44 Urinary tract infections are common in PD but also in MSA, especially in the years before diagnosis. Even though MSA has a shorter prodrome, urinary tract infections are associated with increased risk of MSA when the infection happens 8 years before the appearance of cardinal symptoms. 44 In the general population, urinary tract infections are prevalent, especially in women. However, in PD and in MSA, urinary tract infections affect men and women equally.44,45, 44,45 The reasons for this are not fully understood, and it remains unclear how central or peripheral pathology of synucleinopathies might contribute to increased susceptibility to urinary tract infection, which in turn could exacerbate neurodegenerative processes, as further discussed below. This requires more research, as these infections are linked with the onset of synucleinopathies.
Other epidemiological results are less clear as to when they occur in relation to the prodrome and how they might contribute to synucleinopathies. Some epidemiological studies focused on the presence of pathogen-specific antigens present in the plasma or brain tissue of people with PD by using ELISA, PCR or immunohistochemical methods. Therefore, it is not known if these infections would have occurred before or after the onset of disease. Other studies also relied on self-reporting or used a questionnaire-based methodology to examine potential associations with PD, 46 which can potentially introduce a recall bias and an under- or overestimation of infection.
Nevertheless, from these studies it is apparent that multiple pathogens might increase the risk of developing synucleinopathy or unmask underlying pathogenesis during disease progression. As such, notable links have been established between infections with hepatitis C virus, herpes virus, Helicobacter pylori and other types of pathogens that are further summarized in Table 1.
Prevention and vaccination
Studies have reported that vaccination may decrease the association between influenza or herpes zoster infection and the development of PD after vaccination.46,60, 46,60 However, in one prospective study in people older than 60 years there was no association found between influenza vaccination and risk of PD. 61 Antivirals for the treatment of chronic hepatitis also result in a reduction of associated PD risk. 47,58,59, 47,58,59 Vaccinations against these viruses could potentially reduce the risk of infection and, subsequently, lower the risk of developing PD. Individual factors such as age, overall health, and genetic risk factors could be taken into consideration when making decisions regarding vaccination and to determine the most appropriate vaccination strategy for individuals at risk of developing PD.
Vaccines are available for most of the viruses listed in Table 1; however, vaccination rates remain relatively low, even for people that already have PD. It has been estimated that vaccination coverage for influenza is approximately only half of all PD patients 62 whereas for SARS-CoV-2 vaccination rates in people with PD are even lower than that of the general population. 63 A reason for this could be restricted access for people with PD to primary healthcare 64 but efforts are needed to improve the number of people with PD or related disorders with healthcare access.
Before as well as after diagnosis of synucleinopathies, it could thus be important for people at risk to get vaccinated for several reasons. PD can weaken the immune system, making individuals more susceptible to infections. 65 Infections and illnesses can sometimes exacerbate disease symptoms or trigger temporary worsening. By avoiding illness through vaccination, individuals with PD may manage their symptoms more effectively. Particularly in MSA, or in patients with autonomic dysfunction, patients may be more vulnerable to developing complications from infections such as pneumonia or influenza. Vaccinations against these diseases can reduce the risk of serious complications and hospitalization.
It could thus be hypothesized that infectious pathogens might be associated with an elevated risk of developing a synucleinopathy. However, as we will also discuss in the next section, these associations do not necessarily imply causality, as these observations, although from multiple systematic and observational studies, cannot definitively establish a causal relationship. On the other hand, the association of these risk factors warrant their involvement in synucleinopathies, regardless of the directionality of these associations. Infections could precipitate a preclinical condition and thus accelerate or trigger the onset of synucleinopathy which needs to be further investigated.
A vicious cycle of infections and synucleinopathies?
These historical and epidemiological observations are a good example to illustrate the complexity of pathogen-host interactions: how do we study the relationship between a potential causative agent and a disease that is only diagnosed following a long asymptomatic period? The prodromal phase in synucleinopathies can range from approximately 5–10 years in MSA to 15–20 years in PD.66–69 If infections would act as triggers of disease, they would therefore need to occur around the time when the asymptomatic period would start. An interaction would at best support the contention that an association might exist, but it cannot prove causality.
The protracted prodrome in synucleinopathies now brings us to a second challenge: if an infection happens after the start of the prodrome, how can we be certain about the direction of causation? If an infection happens too close to the time of diagnosis, this would raise concerns about the causative role of the infection since some of the prodromal symptoms can be risk factors for infection. For these cases, the infection might then be a consequence of the disease rather than a trigger of the disease. However, this does not preclude infections from accelerating disease progression or aggravate symptoms.
One of these early cardinal symptoms of PD and MSA, which in some cases manifests during the prodrome, is autonomic dysfunction.68,70–72, 68,70–72 The autonomic nervous system controls several crucial functions such as heart rate, blood pressure, swallowing, digestion, sweating, temperature, or urinary voiding—among others. In PD and especially in MSA, autonomic dysfunction can be severe. Autonomic dysfunction can occur with the loss of peripheral innervation,73,74, 73,74 leading to desensitization.
Dysfunction of the autonomic nervous system can lead to a higher susceptibility to infections via several mechanisms. Swallowing difficulties or dysphagia can lead to aspiration pneumonia, whereas constipation can disrupt the composition of microbiota or lead to fecal retention and an increased risk of urinary tract infections (via translocation of bacteria). Similarly, loss of bladder control, can lead retention of volume in the urinary bladder, which again can increase the risk of urinary tract infections.
Another important function, that is impaired early in MSA, is thermoregulation.75,76, 75,76 Upon infection, an increase in body temperature or sweating would normally indicate signs of infection, but in MSA these functions are impaired. Impaired sweating can create an environment conducive to the growth of bacteria and increase the risk of skin infections in PD and MSA.57,77, 57,77 In the absence of these signs, this can create a dangerous situation where signs of an infection will be muted. Therefore, in some cases, it is possible that the normal signs of infection are missed in which a condition can worsen, causing neurological symptoms, such as confusion or delirium, which require urgent medical treatment.
There is also a more direct way via which the autonomic nervous system is important in the host response against infections. Neurotransmitters of the autonomous nervous system, such as acetylcholine or norepinephrine, can bind to immune cell receptors, which will lead to activation or proliferation of these cells.78,79, 78,79 Alternatively, via the release of neurotransmitters immune cells that are stored in lymphoid organs can be recruited. 80 The loss of autonomic innervation can result in changes in neuroimmune interactions and potentially impair the ability of the immune response to fight off infections. 81
Clearly, due to the protracted nature of synucleinopathies, and their long prodromal phase, there are significant challenges in studying how infections might influence the initiation or the progression of these diseases. Nevertheless, there is growing epidemiological evidence that infections play a role in both disease initiation and disease progression and that infections might have a causative and reciprocal role in the pathogenesis of these diseases.
INFECTIONS ARE POTENTIAL TRIGGERS OF α-SYNUCLEIN TRANSMISSION
Pathogens from the outside environment continuously threaten our body’s defense systems through various routes. Inhalation exposes us to respiratory pathogens, while ingestion introduces harmful agents into our digestive system. Skin contact and compromised barriers allow entry for pathogens, and mucosal surfaces are susceptible to invasion through direct contact. Some pathogens can breach these barriers and enter the peripheral nervous system through direct infiltration or through blood circulation. Some viruses can invade peripheral nerves, or directly cross the blood-brain barrier (BBB) and enter the brain, causing neuropathies, encephalitis, or post-encephalitic parkinsonism.16,30,33,82, 16,30,33,82
The Braak hypothesis postulates that one or two insults occur in the nose or the gut and that this can trigger a cascade of events leading to central pathology in idiopathic PD.83,84, 83,84 In one of their original publications, Hawkes, Braak and Del Tredici mentioned that “a neurotropic pathogen, probably viral, enters the brain via two routes: (i) nasal, with anterograde progression into the temporal lobe; and (ii) gastric, secondary to swallowing of nasal secretions in saliva”. 84 More recently, Borghammer and colleagues further built on this hypothesis and proposed a framework in which the starting point and the anatomical distribution of αSyn pathology could lead to a categorization of PD into distinct subtypes; a brain-first and a body-first subtype, in which pathology specifically related to αSyn progresses via different but predictable routes throughout the body.68,85, 68,85
Although these ideas are focused on PD, there is experimental evidence that the underlying mechanisms related to αSyn transmission are shared with MSA and DLB. 86 Under pathological conditions, αSyn aggregates into small β-sheet-rich assemblies and further aggregate into large filamentous fibrils. 87 When fragmented, fibrillar fragments can transmit and be taken up by adjacent cells. 88 These aggregates can induce the misfolding and aggregation of soluble synuclein proteins in neighboring cells, leading to the progressive spread of αSyn pathology throughout the nervous system. 89
Thus, αSyn aggregates taken up by neighboring cells will serve as templates for the misfolding of soluble αSyn protein and facilitate the transmission of pathology between cells. 89 This has led to the suggestion of a prion-like propagation of αSyn pathology, characterized by adopting abnormal conformations that then propagate pathology through an infectious-like mechanism.90,91, 90,91 While synucleinopathies share similarities with classical prion diseases, it is important to note that the transmission of αSyn between individuals has not been established. The transmissible nature here thus refers to the transmission of αSyn between cells and not individuals.
Several studies have shown that microbial infections can directly trigger the accumulation of αSyn and its conversion from a soluble form to an aggregated state.44,92–94, 44,92–94 This conversion appears to be highly dynamic and occurs within the first hours of infection. 44 The presence of pathogens or their byproducts can alter cellular metabolism and signaling pathways, leading to dysregulation of protein degradation mechanisms such as the ubiquitin-proteasome system and autophagy.95,96, 95,96 Impaired protein clearance can expedite the accumulation of misfolded synuclein proteins and facilitate their aggregation. 97 At any infected site, αSyn is thus prone to accumulate and misfold. It can build up within visceral barrier sites that are exposed to inflammatory pathogens. In this highly reactive and rapidly changing environment post-translational modifications such as phosphorylation, oxidation, acetylation, ubiquitination, glycation, glycosylation, nitration, and truncation can further modify the structural, biochemical, and cellular properties of αSyn. 98
The presence of aggregated αSyn in visceral sites is a observed in all individuals. In normal as well as affected individuals.44,99, 44,99 From these visceral sites, aggregates are potentially prone to travel to the brain via axonal transport. 100 Injection of recombinant αSyn fibrils in peripheral sites leads to progressive symptoms that resemble PD and MSA in animal models.44,101–104, 44,101–104 Successful and predictable experimental transmission of αSyn has now been shown for three common routes to the brain; the nose, the gastrointestinal system, and the urogenital system. 44,101,103–108, 44,101,103–108 Repeated infections or inflammatory exposure within the gut or the urogenital system could thereby predispose to gut-to-brain pathology (body-first type) whereas repeated infections in the nose might predispose to brain-to-gut pathology (brain-first type).109,110, 109,110 Infections in specific peripheral sites could thus contribute to heterogeneity in synucleinopathies as each site can provide a trigger with unique transmission routes and associated pathology.
Other transmission routes, such as systemic transmission have also been studied.111–114 However, the pattern of spread here is less predictable compared to local injections. Nevertheless, given the native role of αSyn in hematopoietic or immune functions, the systemic route might have an important homeostatic role that influences or mitigates the overall burden and the turnover of αSyn protein. Intravenous delivery of αSyn fibrils for instance has been shown to result in pathology that mimics early prodromal stages of PD. 114 It is also not inconceivable that peripheral organs, such as the liver or the kidneys might play a homeostatic role in the clearance of pathological αSyn.115,116, 115,116 Along those lines, systemic transmission could be a relevant and possibly parallel peripheral route via which synucleinopathy could be influenced (Fig. 2).
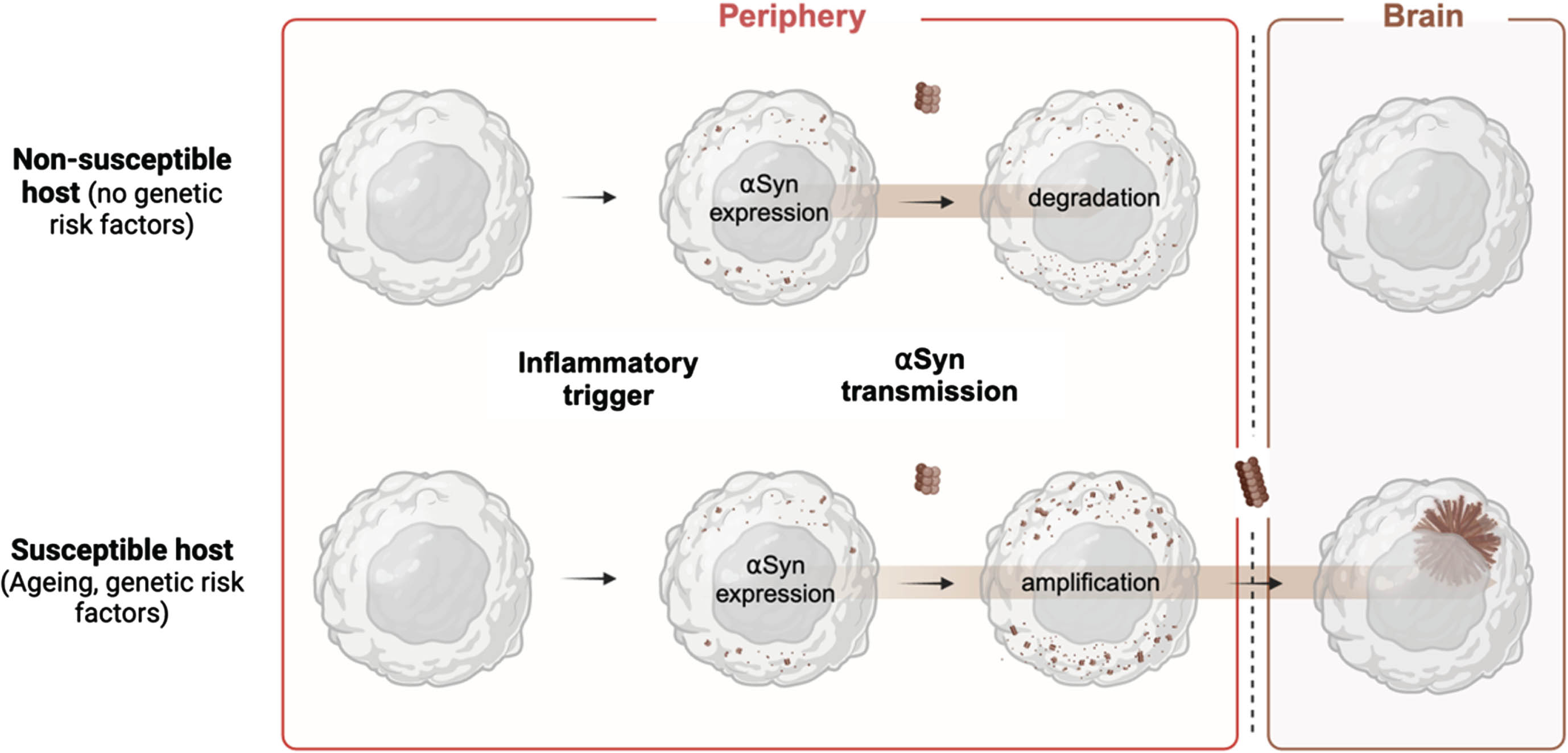
Studying how virulent agents might interact with αSyn in peripheral barrier sites could help us understand how the initial steps of aggregate formation take place. A better understanding of this process might also provide new clues as to how distinct assemblies of are created. Since peripheral aggregates are common in people with and without clinical symptoms, it suggests that some of these aggregates might have only a little seeding capacity. Injection of aggregates isolated from peripheral sympathetic ganglia has indeed shown a lack of seeding capacity compared to aggregates isolated from the brain. 117 Varying efficiencies of transmission could depend on the conformation, the stability, or the maturity of αSyn assemblies. Whether a specific virulent agent may induce the formation of αSyn assemblies with higher seeding capacities may also depend on age at exposure, genetic risk factors and further environmental impacts.
In that regard, it has been shown that distinct types of strains exist in PD, DLB and MSA. 86 Strains of αSyn are fibrils with different molecular structures that each have a specific biological or pathological role because of their unique conformation. While experimental models have provided tentative answers to where and how synucleinopathy might start, they do not yet explain how distinct strains of αSyn might arise. αSyn is known to form different fibril structures with distinct toxicities and seeding capacities. 111 These strains of αSyn are notably distinct in Lewy Body disorders and MSA have as they have a different molecular structure118,119, 118,119. In experimental animal models, fibril strains derived from PD, DLB and MSA patients have unique pathological features, and it shows that a structural-pathological relationship exist between a given strain and the resulting phenotype.118–120
We and others recently postulated that during serial transmission of αSyn pathology the conformation of αSyn can change and that the host cellular environment is responsible for how αSyn assembles and forms strain-specific assemblies.88,121, 88,121 Multiple studies have shown that the formation of strain-specific assemblies from heterogenous smaller assemblies requires multiple steps of structural reorganization, with the rearrangement of intramolecular interactions.86,122–124, 86,122–124 This requires transient interactions with cell-specific chaperones or molecules, or conversely the failure of proteostatic systems, that facilitate the conversion and the maturation of unstable aggregates.125,126, 125,126 This process continues until αSyn aggregates are thermodynamically stable and become the dominant structural conformer. 121 As the transmission of αSyn assemblies from a peripheral trigger site, either from the nose, the gut or elsewhere, is determined by genetic and cellular risk factors,109,127–130, 109,127–130 these risk factors will aid the formation of stable seeds within the cellular environment along the peripheral-central axis in which serial transmission takes place.
Even given the unique genetic makeup and the unique exposome of each individual, the progression of synucleinopathy could thus happen in a way that is unique but still predictable. The conformational landscape of αSyn will gradually and progressively narrow while the disease progresses. The continued and repeated transmission of αSyn will eventually lead to a restricted number of disease-specific strains of αSyn within a larger group of αSyn assemblies.88,121, 88,121 Infectious agents that rapidly trigger aggregation could thus be an important upstream event in the etiology of αSyn strains.
Infections are thus emerging as upstream triggers in αSyn-related idiopathic PD and other synucleinopathies. Insults in the periphery can initiate local pathology and potentially lead to distinct disease subtypes based on synucleinopathy progression routes. Infections rapidly catalyze the accumulation and conversion of αSyn into newly formed aggregates, of which most will be unstable and degraded. Instead, in some cases, these aggregates will persist so that they can transmit to other cells and cause progressive pathology. These peripheral aggregates may propagate to the brain via retrograde axonal transport, as they further mature into disease-relevant assemblies, highlighting the potential and potentially pivotal role of the exposome in the development of synucleinopathies.
MECHANISMS OF HOW INFECTIONS CAN AFFECT SYNUCLEINOPATHIES
Aside from the potential upstream role of infections in the etiology of synucleinopathies, there are several other mechanisms through which infections can influence disease development. This could happen via: 1) direct infection of the brain (in rare cases); 2) an immune response in peripheral organs, including the gut and the bladder, which transmits to the brain via the systemically or via the olfactory epithelium; 3) an alteration of peripheral microbiomes such as in the gut that transmits to the brain; 4) triggering of peripheral αSyn accumulation or aggregations that transmits via the peripheral nervous system; 5) invasion of microbial byproducts into the brain after peripheral infections. In all these scenarios the host immune response to the pathogen could play a central role in the pathology development.
Inflammation is part of the normal immune response to harmful stimuli, which can include pathogens or damaged cells, and is an important contributor to neurodegenerative diseases including synucleinopathies.131–133 Host cells use pattern recognition receptors (PRRs) and other receptors to monitor and identify a wide range of pathogen-associated molecular patterns (PAMPs) derived from different microorganisms including viruses, bacteria, and fungi. 134 They also recognize damage- or danger-associated molecular patterns (DAMPs) released from senescent and dying cells. 135 The activation of the innate immune system mediated by these receptors is emerging as an important pathological mechanism underlying neurodegenerative diseases. 136 There are several families of extracellular and cytosolic PRRs, that include the Toll-like receptors (TLRs), the NOD-like receptors (NLRs) and other cytosolic sensors. PRR activation is the first step of the immune response that triggers specific cellular signaling pathways inducing the production and release of pro-inflammatory mediators (cytokines and chemokines) and the removal of damaged cells (induction of autophagy or cell death).
Inflammatory insults can reach the brain and trigger a highly specialized immune response characteristic of the central nervous system (CNS) or neuroinflammation.137 - 140 This specialized immune response relies on unique immune cells and the lymphatic system that controls an intricate balance between the CNS and peripheral immunity. 141 At the interface of the brain and the periphery microglia and astrocytes work with brain border immune cells in response to inflammatory stimuli. Using different strategies pathogens can trigger a neuroimmune response with long lasting effects on glial activation and cytokines production, with the potential to aggravate an ongoing pathological process. 140 The inflammatory process that occurs during an infection and its proper resolution further depends on the proteostatic cellular machinery. Chronic inflammation or recurrent exposure to inflammatory agents generates stress on these cellular pathways leading to toxic protein accumulation and eventually cell death. This pathological process can be aggravated by aging and the presence of genetic risk factors.
Synucleinopathies are characterized by specific signatures of central and peripheral inflammation. PD is characterized by chronic microglial activation and widespread neuroinflammation.142–145 There is more prominent microglial activation and T cell infiltration in typically affected regions including the substantia nigra.142,146, 142,146 Similarly, MSA is characterized by widespread microglial activation in multiple affected areas. 132 In the case of DLB, microglial activation is early and transient, while in later stages patients lack reactive microglia. Infiltration of natural killer cells and T cells is also observed in these affected areas131,147–149, 131,147–149.Next to central inflammation, signs of peripheral inflammation are also typical of synucleinopathies.150,151, 150,151 Some of these changes include increased proinflammatory cytokines, changes in immune cell numbers and circulating T cells that are reactivated to disease-specific αSyn peptides. 150 In the case of PD, several studies have shown a decrease of circulating lymphocytes and an increase of neutrophils years before diagnosis. 152 These inflammatory changes suggest an early involvement of the immune response in synucleinopathies pathogenesis.
Converging cellular signaling pathways during infection and synucleinopathy
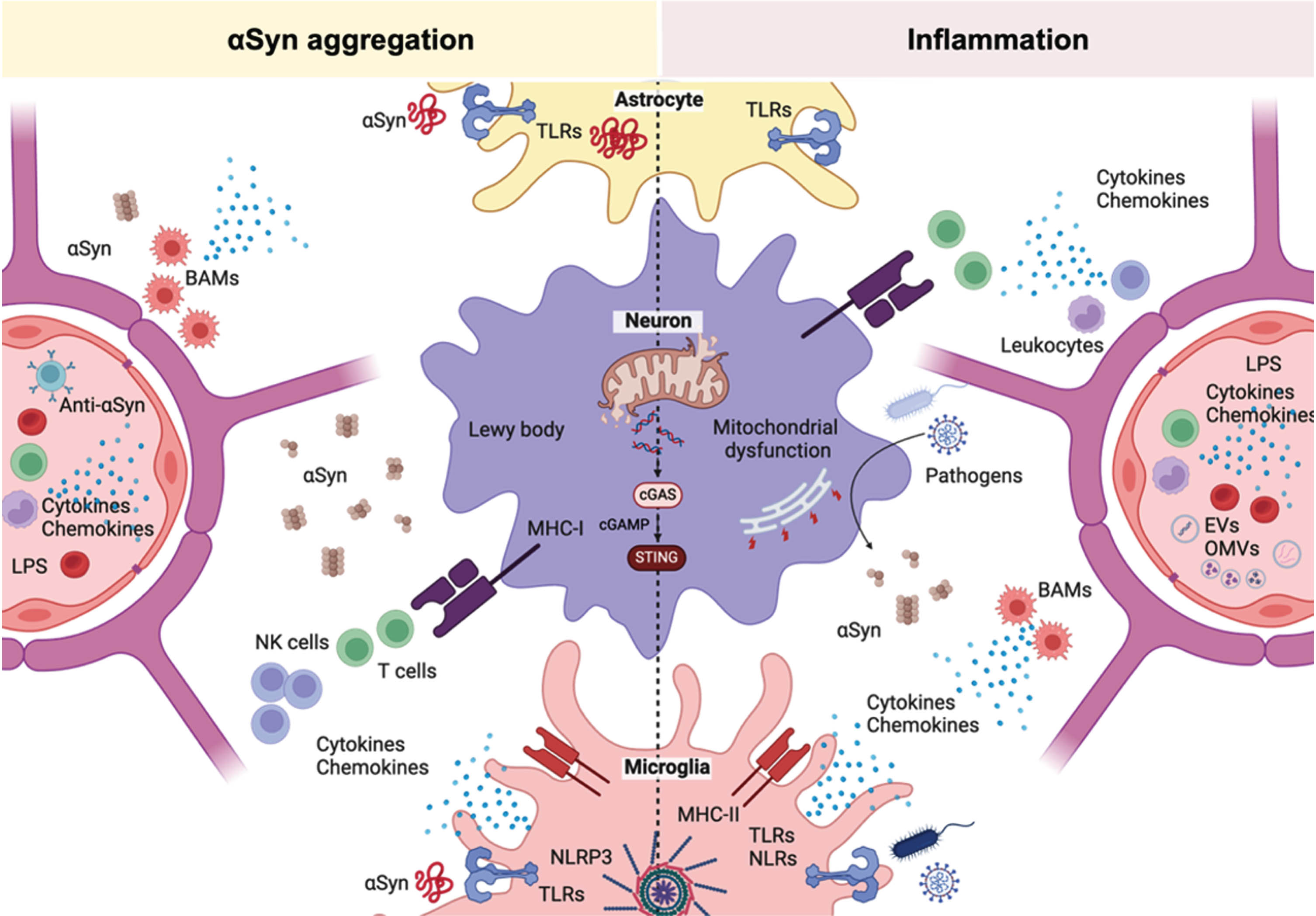
Several cellular signaling pathways have been shown to be affected in synucleinopathies, including impaired proteostasis, vesicular transport and mitochondrial dysfunction. 153 Many of these affected cellular pathways are altered during neuroinflammatory states, where glial cells and neurons respond to a peripheral or central insult. Interestingly, such cellular pathways are also impaired by pathogens during infection. Some pathogens have the capacity to hijack the protein synthesis and autophagy machineries so that they can replicate and avoid the host immune response.82,154, 82,154 Mitochondria are also central to the immune response as these organelles participate in several inflammatory signaling pathways. This includes an innate immune mechanism that detects the presence of cytoplasmic DNA within cells. This pathway plays a crucial role in the body’s defense against infections, particularly viral infections, but it can also sense mitochondrial DNA. This pathway involves cyclic GMP-AMP synthase (cGAS) and the transmembrane protein stimulator of interferon genes (STING). 155 The cellular responses to inflammation can trigger deleterious events, including oxidative stress and cytokine-receptor-mediated apoptosis, which might eventually lead to neuronal loss.
The ability to identify foreign DNA is essential for fending off bacterial and viral infections. Cytosolic DNA is a crucial DAMP, capable of activating the innate immune system through PRRs. Cyclic GMP-AMP synthase (cGAS), is a PRR that can detect foreign DNA from pathogens and self-DNA released during cellular damage, activating stimulator of interferon genes (STING), which will trigger a type I interferon (IFN-I) immune response. In recent years, the cGAS-STING pathway has emerged as a critical player in neurological disorders characterized by chronic neuroinflammation. Viral encephalitis after HSV-1 infection activates the cGAS-STING pathway leading to microglial-mediated IFN-I response. 156 STING-deficient mice are more susceptible to HSV-1 encephalitis after peripheral infection. 156 Moreover, the activation of IFN-I signaling has been observed in postmortem PD human samples and in several mouse models.157,158, 157,158 Impaired mitophagy in PD animal models triggers the activation of the cGAS-STING pathway with functional effects on neuroinflammation, dopaminergic neuron loss and motor function.158,159, 158,159 Inhibition of this signaling pathway alleviates the functional effects and even pathological αSyn accumulation in an αSyn preformed fibrils mouse model. 160
Another innate immune response signaling pathway that is activated upon infection is the inflammasome. Inflammasomes are a family of protein complexes that can sense environmental and cellular danger signals. 161 One of the most studied inflammasome proteins is the NLR family pyrin domain containing 3 (NLRP3)-inflammasome, the apoptosis-associated speck-like protein containing a caspase activating recruitment domain (ASC) and caspase-1. 161 Upon activation, inflammasomes produce and release pro-inflammatory cytokines IL-1β and IL-18 through pyroptotic pores. 161 Markers of activation of this pathway are elevated in the brain and in blood samples from PD.162–164 Activation of the NLRP3-inflammsome pathway is also present in genetic and toxicological mouse models of PD.162,165–167, 162,165–167 Recent studies suggest that downregulation of NLRP3-inflammasome can ameliorate dopaminergic neuron loss and motor deficits in different mouse models of PD. 168
Affected signaling pathways in synucleinopathies resemble those seen as a response to pathogen invasion or inflammatory processes. These changes often involve cellular stress and a combination of upregulation of defense mechanisms and downregulation of pathways essential for cell homeostasis. These similarities suggest that both synucleinopathies and infections may involve common mechanisms of neuronal damage and dysfunction (Fig. 3).
Infections as triggers of synucleinopathy
Viral triggers
As mentioned earlier, there are well-documented cases of viral parkinsonism caused by influenza, coxsackie virus, Japanese encephalitis B, St. Louis, West-Nile and HIV. More recently, there are case reports that infection with SARS-CoV-2 causes L-DOPA responsive parkinsonism.41,169, 41,169 The reason why these viruses cause parkinsonism is not well understood and whether viral invasion of the CNS is necessary for the development of neurological symptoms is still under investigation.
However, many of these viruses have been shown to preferentially target the midbrain and the dopaminergic system.41,170,171, 41,170,171 These viruses have the potential to access the CNS via different mechanisms. 172 Some of these pathogens can access the CNS from the olfactory epithelium or alternatively, via innervation from visceral organs. Others access the brain by infecting immune cells that transport the virus over the BBB into the CNS. Another mechanism to enter the brain is by extravasation from brain capillaries. Viral infection can decrease the expression of tight junction proteins and increase vascular permeability to facilitate this process. 173 After entering the CNS, infectious pathogens can trigger a neuroimmune response, which includes glial activation, cytokine and chemokines release, oxidative stress, peripheral immune cells infiltration and potentially αSyn aggregation. 168
Recent evidence has shown that several viruses can cause αSyn pathology. Brain infections with influenza virus, West-Nile virus, picornavirus, coxsackievirus or SARS-CoV-2 have been shown to trigger αSyn pathology.157,174–178, 157,174–178 Infection with H1N1 via the olfactory route inhibits proteostasis in the substantia nigra of immunocompromised Rag KO mice and leads to αSyn aggregation in vivo. 177 Coxsackievirus can directly cause aggregation of αSyn in cell and animal models and again cause Lewy-like pathology in vivo. 174
Similarly, in experimental models of SARS-CoV-2 infection, intracellular αSyn aggregates in the substantia nigra were found in SARS-CoV-2 infected macaques. 176 SARS-CoV-2 is generally not a neurotropic virus, but we and others demonstrated that intranasal infection in hamsters can cause persistent inflammation in the olfactory and cortical regions,179,180, 179,180 followed by tau and αSyn pathology, 180 in the absence of overt CNS invasion.
Microglial activation and cytokine production in the brain can persist long after SARS-CoV-2 infection has resolved, suggesting that CNS damage after infection may be due to a cytokine storm and thus the neuroimmune response rather than direct infection with SARS-CoV-2.181,182, 181,182 Further studies on the time course in intranasally infected hamsters revealed that microglia cell density and αSyn immunoreactivity decreased at 6 days post SARS-CoV-2 infection, then rebounded to overt accumulation at 21 days post infection. This biphasic response was most pronounced in the amygdala and striatum, brain regions affected in PD, and in female hamsters. Importantly, female sex appears to predispose to long term complications after SARS-CoV-2 termed post-COVID19. 178
Thus, SARS-CoV-2 profoundly disrupts brain homeostasis without neuroinvasion, via neuroinflammatory and protein regulation mechanisms that persist beyond viral clearance, providing a potential link between viral infection and the risk of neurodegenerative diseases.
In vitro experiments have shown that SARS-CoV-2 peptides derived from the SARS-CoV-2 spike protein have amyloidogenic properties that seed αSyn fibrillization.183–186 Furthermore, the spike protein primed and activated the NLRP3 inflammasome in human microglia, and enhanced αSyn-mediated NLRP3 activation in microglia cells. 187 There is evidence that these viral proteins still cross the BBB and reach the brain parynchema.188,189, 188,189 Of note, the neurovascular unit is altered upon SARS-CoV-2 infection. After infection there is brain damage to cerebral small vessels evidenced by increased string vessels and microvascular pathology, 190 as well as pericyte-mediated capillary constriction. 190 Under viral infection-induced BBB alterations and peripheral inflammation, viral proteins may cross into brain parenchyma causing neuroinflammation and concomitant neuronal damage.
However, such mechanisms are thus not restricted to the CNS or the nose-brain axis. Indeed, as mentioned earlier, αSyn aggregation is also observed in peripheral tissues, including the gut, the skin, and the urinary bladder. Infections can trigger αSyn aggregation and its release from neutrophils. 44 Molecular dynamics simulations suggest that a SARS-COV-2 spike protein fragment, cleaved by neutrophil elastase under inflammatory conditions, favors αSyn fibrillar seeding conformations, suggesting again that peripheral immune cells could potentially contribute to αSyn aggregation during infections.184,191, 184,191
Gut triggers
The involvement of the gut in synucleinopathies has been a field of intensive research. Gut symptoms such as bloating and constipation appear early in some people with PD, indicating that the gut might be a starting site of pathology for these cases. This is in conjunction with early observations of αSyn pathology of the enteric nervous system as well as autonomous nervous system.68,110,192, 68,110,192 There is a bidirectional connection between the gastrointestinal tract and the brain, called the gut-brain axis, and it can be influenced by gut infections, dysbiosis, and inflammatory responses.
Changes in the gut microbiota happen early in PD. Multiple studies have shown that intestinal microbiota dysbiosis can be a prominent feature.192–195 Several factors can influence the gut microbiome, such as geographic factors and ethnicity. However, despite these factors, PD related dysbiosis is broadly characterized by a lower relative abundance of anti-inflammatory bacterial genera and an increase of pro-inflammatory and opportunistic pathogens.195,196, 195,196 These can include Escherichia, Klebsiella, and Porphyromonas.195,196, 195,196 Even though the causes for dysbiosis in PD are not fully understood, the gut microbiota can be affected by exposure to pollutants, pesticides, and infections. 197 Research in preclinical PD models has shown that gut microbiota manipulation can influence symptoms and pathology including αSyn aggregation.198–200 More recently, it was shown that a fecal microbiota transplantation can potentially alleviate motor symptoms in early-stage PD patients. 201
Gut dysbiosis and infections can cause pathological changes in the immunoregulatory function of the gut and increase intestinal permeability. Increased intestinal permeability is a characteristic of PD. 202 It is accompanied by changes in gut microbial metabolites, such as bacterial lipopolysaccharides (LPS) and short-chain fatty acids (SCFAs). Inflammatory mediators can be released from commensal and pathogenic gut microbes and trigger unwanted systemic and central effects. It has been proposed that the disruption of the gut epithelium could trigger a feedback loop that alters the microbiome to a more pro-inflammatory profile resulting in an increased permeability, oxidative stress, and aggregation of αSyn in the gut. 203
Experimental and clinical studies have implicated the enteroendocrine cells of the gut epithelium.204,205, 204,205 Enteroendocrine cells can sense the luminal content in the gastrointestinal epithelium, and they play crucial roles in regulating gastrointestinal functions, including digestion, nutrient absorption, and gut motility by producing and releasing hormones. LPS and SCFAs are potent activators of enteroendocrine cells and interestingly, enteroendocrine cells also produce αSyn. In response to inflammatory stimuli, these cells can transiently express and release αSyn.204,206, 204,206 This thus directly links infectious agents and microbial metabolites (LPS or SCFAs) with the production of αSyn in the gut (Fig. 4).
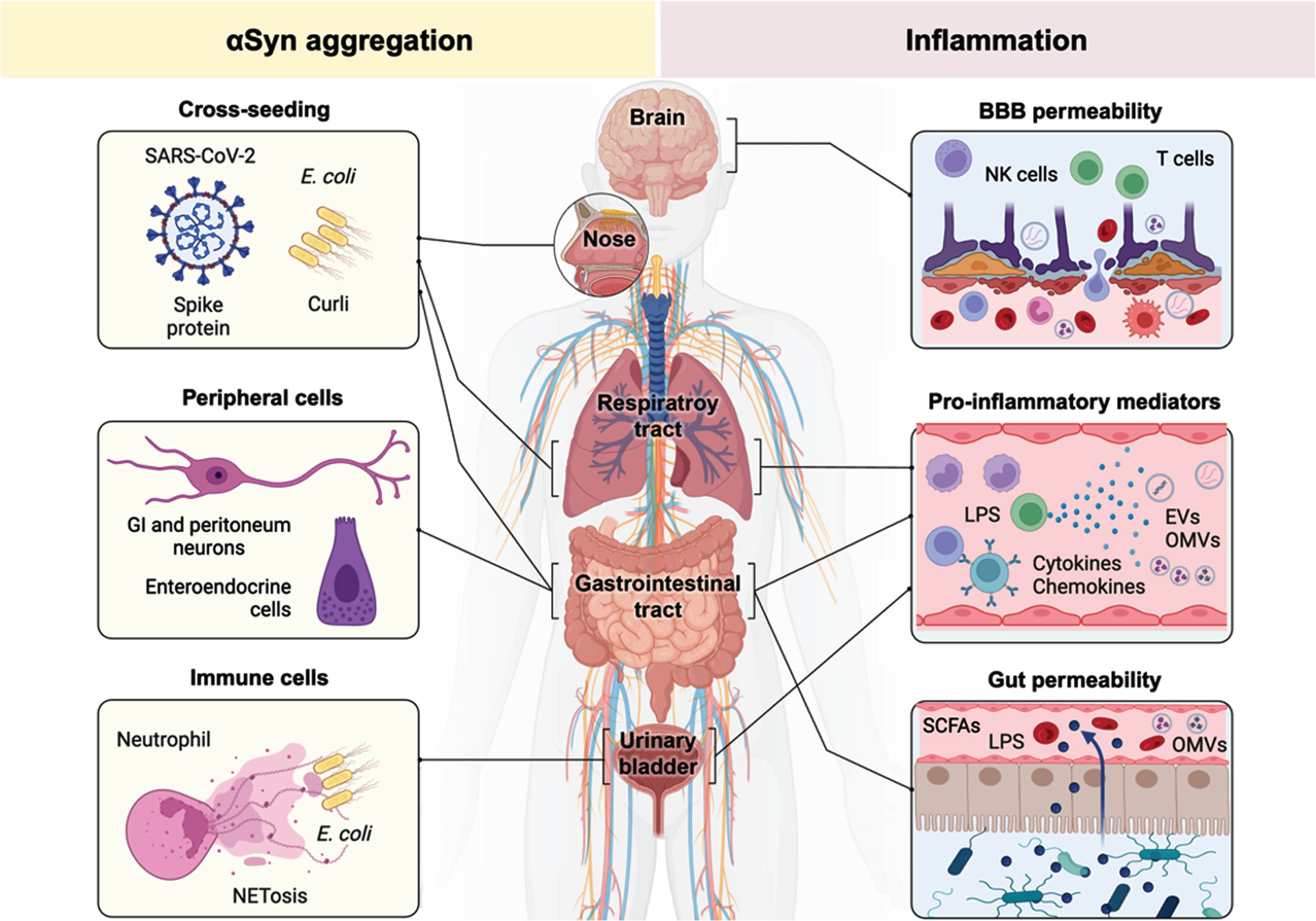
Microbiota-derived SCFAs are common bacterial fermentation products. These gut microbial metabolites, such as for instance butyrate, propionate, or acetate, have many physiological functions including reducing inflammation and preserving epithelial integrity. 207 SCFAs-producing bacteria are less abundant, and these metabolites are decreased in the feces of PD patients.208–210 On the other hand, the release of SCFAs from the gut can influence microglial homeostasis. 211 Animal studies have demonstrated that SCFAs have functional consequences by increasing αSyn aggregation and the development of motor dysfunction. 212
LPS is a component of the outer membrane of Gram-negative bacteria and a potent activator of the innate immune response. LPS from the Enterobacteriaceae family is one of the most immunogenic forms and is found abundantly in the gut of PD patients.196,213, 196,213 It has been recently shown that PD patients have elevated levels of LPS in the blood and high levels of LPS binding protein (LBP) during the prodromal phase are associated with an increased risk of PD.214–216 LPS activates the innate Toll-like receptors 2 and 4 and triggers an immune response that promotes the production of pro-inflammatory cytokines, chemokines, and reactive oxygen species. TLRs activation in microglia leads to the production of pro-inflammatory cytokines, the initiation of the adaptive immune response and recruitment of T and B cells. In PD models, LPS induces a neuroinflammatory response that ultimately leads to neurodegeneration.217–220 This is likely caused by the selective vulnerability of dopaminergic neurons of the substantia nigra to LPS insults.217–220 Additionally, LPS administration can trigger αSyn expression and aggregation.221–224 This has been observed in both the gut as well as in the brain, and more specifically in the substantia nigra, of animal models. 221–224
Aside from the direct or cis-acting pathogenic effects of bacteria on αSyn and PD pathogenesis, bacteria can also exert trans-acting effects. This happens via the release of outer membrane vesicles (OMVs) or alternatively, via the release of amyloidogenic proteins. Gram-negative bacteria can release OMVs from the stomach or the gut which can cross the BBB. 225 E coli or Helicobacter pylori OMVs are only a size of 20–200 nm and thus can readily cross the BBB to infect neurons and glial cells. 226 When taken up by astrocytes, this will lead to the expression of complement proteins and cause an inflammatory reaction. 227 OMVs loaded with LPS can also interact with microglia and other PRRs-expressing cells. 225 Some of these bacterial vesicles also transfer different bacterial molecules such as RNAs, endotoxins and toxins. 225 Because of the direct transfer of pathogen metabolites to the cytosol, OMVs can trigger the activation of the intracellular NLRP3 inflammasome pathway resulting in the release of pro-inflammatory cytokines and mitochondrialdysfunction. 228
Next to these potentially pathogenic carrier vesicles, bacteria can release amyloidogenic proteins. One such well-characterized bacterial protein is the amyloid protein curli. Curli is produced by E. coli and the protein can aggregate into fibrils to facilitate biofilm formation and help with cellular adhesion. Curli fibrils play dual roles in promoting bacterial colonization and infection while also triggering the host immune response, contributing to both bacterial persistence and host defense mechanisms. Curli can cross-seed the aggregation of αSyn in vitro and injection of curli fibrils can lead to αSyn pathology in the gut. 229 Pathological transmission of αSyn to the CNS has been observed in mice after transplantation of curli-expressing E. coli. 230
Aside from gut bacteria, viral gut pathogens have also been associated with synucleinopathy. 231 Viral infection with norovirus can induce a persistent increase in αSyn expression in the enteric nervous system. 92 Here, the degree and the duration of viral infection in infected cases correlates with the expression of αSyn in gut biopsies. 92
Bacterial and viral gut infections can thus have prominent effects on the gut and the brain. They influence gut permeability resulting in systemic inflammation and neuroinflammation. Via the gut-brain axis gut infections can influence brain health and act as a trigger of synucleinopathy.
THE ROLE OF α-SYNUCLEIN DURING THE HOST RESPONSE AGAINST INFECTIONS
Point missense mutations in the SNCA locus but also gene duplication and triplication cause autosomal dominant PD, indicating that αSyn plays a central role in familial PD. 232 SNCA is also a risk factor for idiopathic PD as non-coding variants are associated with an increased risk of PD or DLB. 233 αSyn is mostly studied for its role as a synaptic protein in neurons but new and compelling evidence is emerging that αSyn is also involved in immune function.
Several studies have now shown that αSyn has an important role in the innate and adaptive immune response.5,234, 5,234 αSyn expression is carefully controlled during infections.92–94 The way this happens is not fully understood, but the presence of inflammation-related regulatory regions in the promoter of αSyn could regulate its expression during inflammation. The human SNCA promoter includes a 34 kb upstream region that contains several additional regulatory regions including the interferon- or other cytokine responsive elements of STAT1 and NF1. 235 These regulatory elements in the SNCA promoter could thus be transcriptionally activated by pro-inflammatory cytokines such as interferon-γ, IL-6 or growth factors that can cross theBBB.236,237, 236,237
The role of αSyn in the innate immune response
Innate immunity involves the recognition of specific ligands by PRRs and the production of inflammatory mediators to limit infections and initiate tissue repair. TLRs are expressed by immune and non-immune cells, including microglia, neurons, astrocytes, and oligodendrocytes. 238 TLRs recognize a wide range of PAMPs 154 and DAMPs, including αSyn. 135
Increasing evidence thus suggests the involvement of TLRs in the pathogenesis of synucleinopathies. 239 It has been reported that in postmortem PD brain tissue there is an increased expression of the microbial-associated molecular pattern (MAMPs) receptors, TLR4 and TLR2.240,241, 240,241 In neuronal cell cultures, TLR2 activation increases αSyn protein levels. Since αSyn is recognized by TLRs, it could serve as a chemoattractant for the recruitment of T and B cells. 92 In PD patients, neurons with αSyn pathology have increased expression of TLR2 241 , which could make them particularly susceptible to the pro-inflammatory effects of pathogen derived MAMPs. Moreover, αSyn can increase the expression of TLRs in microglia to promote neuroinflammation, 242 Different high molecular weight assemblies of αSyn have varying affinities for TLRs and also activate the immune response with varyingefficiencies.243–245
Besides the effects of αSyn on microglia, αSyn also modulates the astroglia immune response. The intracellular accumulation of αSyn in astrocytes is necessary for the induction of a pro-inflammatory response. While astrocytes internalize αSyn mediated exclusively by TLR2, pro-inflammatory cytokine expression can be mediated by both TLR2 and TLR4.245–247 These observations have led to the hypothesis that astroglial TLR2 could play a dual role in synucleinopathies. On one hand by protecting neurons by the internalization of potentially toxic extracellular αSyn, and on the other hand by triggering neuroinflammation. 238
In the periphery, αSyn has similar effects on leukocytes via stimulation of TLRs. αSyn stimulates the maturation and activation of dendritic cells through TLR4. 92 In mice, αSyn is required for both a normal pro-inflammatory response to bacterial peptidoglycan and the T cell mediated response to immunization in the peritoneal cavity. 248 Interestingly, neurons that innervate the peritoneum seem to be the source of αSyn as a ligand for TLRs, supporting a mechanistic link between neuronal pathology and the immune response in theperiphery.
The role of αSyn in the adaptive immune response
Adaptive immunity requires the activation of antigen-sensitized cytotoxic T cells and release of cytokines and chemokines in response to foreign molecules or microorganisms. While T cells help to eliminate pathogen infected cells, B cells produce antibodies to protect against extracellular microbes and toxic molecules. 249 T cells can only recognize antigenic peptides displayed by major histocompatibility complex (MHC)-I or MHC-II molecules on the cell surface 249
MHC-I molecules are expressed in all cells to present antigen peptides to CD8 + T cells, which allows them to identify and eliminate infected cells. 249 Under normal conditions CD8 + T cells are tolerant to autologous or self-proteins. However, during infections cells can express microbial genes and present these ‘non-self’ antigenic peptides allowing CD8 + T cells to eliminate these cells. Nigral dopaminergic neurons express MHC-I suggesting that these neurons are potentially susceptible to T cell mediated cytotoxicity 250 . T cell infiltration in areas of the brain with high αSyn burden is seen in PD and DLB patients as well as in the brain of synucleinopathy animal models.147,251–253, 147,251–253 This reinforces the idea that the adaptive immune response is a potential driver of pathogenesis. Furthermore, the infiltration of cytotoxic CD8 + T cells has been seen as an early pathological event that precedes αSyn aggregation and neurodegeneration in the substantianigra. 254
While MHC-I molecules are expressed ubiquitously, MHC-II molecules are expressed on immune antigen presenting cells. MHC-II molecules present peptides from endocytosed molecules to CD4 + T cells to activate them. 249 Genome-wide association studies (GWAS) have found an association between PD risk and the human leukocyte antigen isotype DR (HLA-DR) alleles. HLA-DR is a cell receptor heterodimer expressed on the surface of antigen presenting cells including microglia and macrophages. Reactive microglia and border-associated macrophages positive for HLA-DR are abundant in the substantia nigra of PD cases.143,255,256, 143,255,256 HLA-DR are part of the MHC-II family, and the genetic link between HLA-DR variants and an increased risk of developing PD supports a role of the adaptive immune response in PD pathology.257–259
Within these lines, circulating T cells from PD patients recognize specific αSyn peptides. 260 Pathological αSyn can trigger an immune response and inflammation, producing autoreactive T cells. 261 This loss of self-tolerance could be associated with environmental factors including infections. 262 Here, repeated exposure to αSyn in the context of infection could thereby cause cellular damage. Furthermore, the HLA-DR allele HLA-DRB1*15 : 01 has been linked to an increased risk of PD, and this allele may also be involved in the presentation of αSyn peptides to T cells. 263 Peripherally circulating CD4 + and CD8 + T cells derived from PD patients produce cytokines in response to αSyn, suggesting there may be a chronic memory T cell response in PD. 264 MHC-II expression and T cell infiltration are essential for neuroinflammation and loss of nigral dopaminergic neurons in a mouse model of αSyn overexpression. 265
It is known that infections that activate the immune system may exacerbate existing conditions or trigger new disease activity in autoimmune diseases. For example, urinary tract infections or respiratory tract infections, which are common, have been associated with increased risk of relapses in multiple sclerosis.266,267, 266,267 The immune response to these infections might lead to increased activity of autoreactive T cells that attack the central nervous system. In the case of PD, and given the potential role of infections in synucleinopathies, peripheral infections could trigger autoimmune activation and cause αSyn autoreactive immune cells to attack αSyn presenting cells and contribute to neurodegeneration via a similar mechanism although it needs to be investigated if such links might indeed exist.
An emerging role for the immune response at the brain border
CNS or border-associated macrophages comprise perivascular, meningeal, and choroid plexus macrophages with phagocytic capacity, which support the BBB by scavenging harmful molecules and promoting efficient immune surveillance, including antigen presentation and cytokine production.268,269, 268,269 Leptomeningeal macrophages migrate postnatally into the perivascular space and differentiate into perivascular macrophages. 270 As described above, under disease conditions with alterations of the BBB, such as in synucleinopathies or neuroinflammation 271 peripheral immune cells potentially invade the CNS and encounter the response of border-associated macrophages.272,273, 272,273 Furthermore, studies in PD models indicate that peripheral monocyte infiltration plays a critical role in αSyn-induced neuroinflammation and neurodegeneration. 274
There are only few studies which address the role of border-associated macrophages as potential gatekeepers in such CNS immune cell invasion. A recent study in a model of synucleinopathy has shown that border-associated macrophages, not microglia, are responsible for CD4 + T cell antigen presentation and recruitment necessary for tissue infiltration and cytokine production upon αSyn pathology. 255 Interestingly, αSyn oligomers were detected in perivascular macrophages in synucleinopathy brain tissue. 275 Depletion of border-associated macrophages prevented αSyn-induced neuroinflammation, including microglial reactivity, T cell infiltration, and monocyte recruitment. 255 Whether this has therapeutic implications for synucleinopathies requires further research. 276
If T-cell and CNS macrophage responses are associated with αSyn pathology in synucleinopathies, then infections that cause neuroinflammation could further exacerbate these processes and thereby neurodegeneration. Proliferation of perivascular macrophages was shown to contribute to the development of encephalitic lesions during simian immunodeficiency virus (SIV) infection of adult macaques. 277 Interestingly, border-associated macrophages but not disease-associated microglia show overt long-term transcriptional alterations which persist after resolution of trypanosoma brucei infection in mice. 278 Thus, single infections may alter the brain’s resident immune response at the border. Border-associated macrophages also play a critical role in protecting the brain parenchyma from viruses. In mice infected with lymphocytic choriomeningitis virus (LCMV), depletion of border-associated macrophages prior to infection or conditional deletion of interferon-receptor signaling pathways in myeloid cells resulted in extensive viral spread into the CNS. This appeared to be mediated by MHC-II-positive meningeal macrophages, and low numbers of these cells, as seen upon LPS challenge, correlated with higher viral load. 279
Altogether, available evidence supports the involvement of brain resident and peripheral immune cells driving pathological events that can lead to neurodegeneration in synucleinopathies. Many of these cellular responses could potentially be triggered by infections and further aggravated by genetic risk factors.
INFECTIONS INTERACT WITH GENETIC RISK FACTORS OF SYNUCLEINOPATHIES
Within the hypothesis that infections might be involved in the etiology of sporadic forms of synucleinopathy, an important role has been proposed for genetic risk factors as ‘facilitators’ of disease. 280 Certain pathogens possibly initiate or exacerbate the neurodegenerative process but in most individuals that are exposed to pathogens, infection will not lead to the onset of disease. It is likely that in most cases, infections will not cause neurological disease as research suggests that infections and sustained pathological inflammation needs to interact with genetic vulnerabilities for synucleinopathy to develop.5,280, 5,280 (Fig. 1). The low penetrance and heritability of most PD or DLB-linked variants, suggest that cumulative exposure to environmental factors such as inflammatory toxins, pesticides or infections may be necessary to reach a level of cellular stress where these genetic factors become physiological relevant and lead to neurodegeneration.
Infections can trigger the immune system to release inflammatory molecules that can infiltrate the central nervous system, leading to neuroinflammation. Overlapping affected signaling pathways between neuroinflammation and neurodegenerative diseases reinforce the notion of a potentially positive feedback loop leading to cellular stress and dysfunction. Multiple PD-associated genes encode proteins with roles in the regulation of cellular transport and vesicle function, critical for the development of the immune response.281,282, 281,282 PD-linked variants in mitochondrial genes also affect mitochondrial function important for the elimination of pathogens and danger signaling during infections. It is possible that genetic risk factors could gain physiological relevance under this cellular stress condition, further amplifying the pathological process. Environmental factors such as bacterial or viral infections but also gut dysbiosis or exposure to toxins may interact with genetic susceptibility in predisposed individuals to influence disease risk.
The relationship between specific PD risk genes and susceptibility to infections is an area of ongoing research. While only a small fraction of genes are implicated in familial PD, there is a myriad of gene variants that potentially increase the susceptibility to develop idiopathic PD. These risk factors are likely upstream in the disease process and many of these genes are implicated in immune function and neuroinflammation. Variants in several of these genes (SNCA, LRRK2, GBA, PRKN, PINK1, etc.) could potentially affect our response to infections and modulate inflammatory processes, which may in turn influence the risk of developing PD.
On the other hand, some genetic variants associated with PD may confer protection against infections or modulate the immune response in beneficial ways. Variants in genes involved in innate immunity or pathogen recognition may enhance the body’s ability to combat infections.
During infections, LRRK2 PD variants can modulate the susceptibility to different infections.283 - 285 For example, LRRK2 G2019S, one of the most common risk variants of PD, is known to potentially protect against respiratory infections by modulating bacterial lung infection. 286 It therefore seems that bacterial infections in the lung (where LRRK2 is highly expressed) could interact with LRRK2 risk, but whether other types of infections might as well be associated with this particular risk factor is unknown. It will be important for future studies to examine whether such unique host-pathogens interactions might exist. Genetic risk factors may modulate the onset or the course of PD in a manner that depends on the type of pathogen. 280 Knowing which risk variants might interact with which type of pathogen could aid in the prevention and diagnosis of different subtypes of PD or other synucleinopathies.
In the case of αSyn, several experimental studies have shown that SNCA deletion results in an increased susceptibility to West Nile virus, Venezuelan equine encephalitis, reovirus and Salmonella typhimurium infections.93,94, 93,94 It has been proposed that this enhanced susceptibility could be due to the role of αSyn in modulating proteostasis and interferon signaling and that αSyn has an important role on the host immune response during infection.94,157, 94,157
Based on the biological classification of PD,287,288, 287,288 the development of some subtypes of PD could rely on a unique set of virulent interactions. On the one hand, pathogens can cause the clinical manifestations of PD, but in the absence of αSyn pathology (Fig. 1). These sporadic cases are distinct from the post-encephalitic cases of parkinsonism, but they do trigger dopamine-specific neurodegeneration after exposure to a specific pathogen. 280 Experimental models have shown that in the presence of PD risk factors, gut infections can directly lead to dopaminergic degeneration, even in the absence of αSyn pathology.289,290, 289,290
On the other hand, infections can directly trigger αSyn aggregation and lead to the deposition of αSyn in peripheral sites.44,248,291,292, 44,248,291,292 αSyn could then be prone to spread from these sites and trigger pathology in the CNS via connected regions. However, despite the presence of Lewy bodies, these types of PD might be fundamentally different and therefore require a different preventive or therapeutic approach due to distinct underlying biological mechanisms.
For instance, for the same risk factors PD patients can be either negative or positive for αSyn pathology. Mutations in LRRK2 are some of the most common PD-linked genetic risks and contribute to both genetic and sporadic forms of the disease.293–295 Some LRRK2 carriers with idiopathic PD have no detectable αSyn pathology, whereas other LRRK2 carriers are positive for αSyn pathology (Fig. 1). Accumulating evidence suggests that LRRK2 is involved in inflammation and the immune response. 296 LRRK2 is expressed in high levels in immune cells297–300 and can modulate pro-inflammatory cytokine production. 301 The cellular function of LRRK2 is important for many cellular processes including vesicular trafficking, autophagy, and lysosomal function 300 .
The appearance of PD symptoms in the absence of αSyn pathology has important implications, as it has been debated if αSyn is required for the clinical diagnosis of PD. αSyn is a cardinal feature in PD and other synucleinopathies, but in some cases pathological αSyn aggregates are undetectable via diagnostic assays or even in the brain. For those patients, PD is likely not a synucleinopathy and the mechanism of disease and the biology of disease would be fundamentally different.
Such examples can also be found in cases with mutations in the mitochondrial genes PINK1 and Parkin 106 . The phenotype in these patients could as well be linked to infections as these genes have been associated with increased susceptibility to intracellular pathogens.302,303, 302,303 Parkin plays a key role in the innate immune response to intracellular infections by targeting them for ubiquitin-mediated autophagy in a similar role that this protein plays in mitophagy. 304 PINK1 is functionally implicated in the response to gut bacterial infection in PD models, where Pink1 deficient mice develop motor deficit after infection 290 . It has been proposed that in the absence of Pink1 mitochondria specific CD8 + T cells can contribute to the degeneration of dopaminergic neurons 290 , adding to the evidence for a potential autoimmune mechanism in Pink1/Parkin-related PD.
Only little is known about how the underlying biology of PD and an individual’s clinical presentation are potentially linked with the exposome. It is possible that exposure to different pathogens can modulate differential disease outcomes in people with the same genetic risk. Future research will need to investigate how these pathological interactions are mediated and can be prevented. In addition, research will need to carefully assess the patient’s exposome in the context of their genetic risk.
Based on the available epidemiological studies we suggest that microbial pathogens can be a precipitating factor in the etiology of PD. Except maybe for very rare cases, virulent pathogens do not directly cause PD, but they are a potential upstream determinant in the disease pathway. Certain types of pathogens might uniquely interact with genetic risk in predisposed individuals. In the case of DLB and MSA only little is known about the types of pathogens that could trigger disease, as epidemiological studies about infectious pathogens in DLB and MSA are scarce. Nevertheless, infections can have lasting effects on the brain, via direct or systemic routes, and chronic effects can outlast the acute effects of the initial infection.
FUTURE DIRECTIONS
Implications for treatment, diagnosis, and prevention
PD is the second most common and the fastest growing neurodegenerative disease affecting more than 4 million people worldwide and its prevalence has doubled in the last 25 years. 67 This increase persists even after correcting for aging suggesting that changing environmental factors could be important players in the pathophysiology of PD. Here, attention has been given to the role of pesticides, but less attention has been given to the role of other inflammatory agents, such as microbial pathogens.
More recently, the neurological symptoms observed after SARS-CoV-2 infection have brought new attention to the long-term neuroinflammatory effects on brain health after infection. The evidence reviewed here suggests that infections could have a role in the etiology of synucleinopathies (Fig. 1). Infections are part of the exposome, and this includes the total environmental exposures that individuals encounter throughout their lives, including factors such as pesticides, heavy metals, air pollution, solvents, and dietary habits. In PD and related synucleinopathies, these exposures may play a role in disease onset or disease development. 305 Studies have linked pesticides, herbicides like paraquat and rotenone, heavy metals such as lead and manganese, air pollution, and certain industrial chemicals (trichloroethylene, perchloroethylene or polychlorinated biphenyls) to an increased risk of PD.305,306, 305,306 When this is further accompanied by aging and immunosenescence (the inability of the immune system to appropriately respond to infection), exposure to environmental factors will lead to increased susceptibility and cause mild or chronic neuroinflammation. 307 Understanding the contribution of different environmental factors is thus crucial for developing preventive strategies and identifying modifiable risk factors for PD.
Because of the clinical heterogeneity in synucleinopathies, subcategorization or stratification of at-risk patients will be key to achieve better therapeutic results. 308 Even though in recent years great progress has been made in the development of molecular diagnosis for synucleinopathies, including protein seeding amplification assays, skin biopsies and new imaging modalities, there are no early diagnostic tests or disease progression markers that could aid in diagnosis or taking preventive measure in the case of modifiable genetic risk factors. Identifying individual risk factors, genetic or environmental, is imperative to perform personalized risk assessment and enable early intervention. It would allow preventive measures to be taken such as lifestyle intervention, diet, exercise or even medication to mitigate risk factors. By understanding an individual’s risk, preventive therapies could have the potential to delay disease onset or improve disease outcomes for those who are at risk of developing PD.
It will also be important to study and consider stratification to evaluate the efficacy of preventive measures. For example, despite the abundant evidence of the link between influenza infections and PD, a recent study based in the UK and Finland found no association with reduced risk of PD and influenza vaccination after 12 years, although a significant association was found between influenza infection and other types of dementia. 43 This suggests that specific host-pathogen interactions might be important in neurological disorders where co-pathologies are present. Recognizing familial or genetic risk factors is vital for making informed decisions about vaccination.
Understanding these risks helps tailor recommendations and emphasizes the importance of vaccination in protecting against potential health threats, especially for those who are predisposed. A downstream consequence of peripheral or systemic infection is neuroinflammation. 309 Neuroinflammatory changes can remain present in the brain long after resolution of infection or even in cases where the pathogen cannot be detected in the brain.181,182,310, 181,182,310 It has been suggested that the neurodegenerative process is driven by a long lasting low-grade neuroimmune response rather than by direct viral neuroinvasion.181,182,310, 181,182,310 Persistent neuroinflammation could help the brain to protect itself from subsequent insults via a primed innate immune system, but at the same time it will aggravate the age-related pathological process.
Much effort has been put into developing therapies that efficiently target neuroinflammation. Several of these approaches include anti-inflammatory medication or the targeted modulation of microglial activity. Another approach is to inhibit the activation of the inflammasome. 311 Here, several compounds are being studied for their potential to inhibit inflammasome activation directly. 312 These include small molecule inhibitors targeting specific inflammasome components.311,312, 311,312 Immunomodulatory therapies, including monoclonal antibodies aim to target specific immune cells or cytokines, which are also being investigated to regulate the immune response 313 . Lifestyle factors, such as diet, exercise, and stress reduction techniques, may help reduce inflammation and oxidative stress since they have been shown to ameliorate motor- and non-motor symptoms in PD.314–319
The recent and increasing popularity of GLP-1 receptor agonists, which are currently used for the treatment of type-2 diabetes, could also help to reduce inflammation in synucleinopathies. GLP-1 receptor agonists are being studied for the treatment of PD as they are potent inhibitors of neuroinflammation via inhibiting NF-kB activation in microglia.320,321, 320,321 In turn, this reduces the conversion of astrocytes into neurotoxic astrocytes. 320 Recent clinical studies have shown that the progression of motor and non-motor symptoms is slowed down or can even be stabilized when the GLP-1 receptor agonists are taken early in the disease course.320–322 While larger trials are ongoing, much research is needed to further evaluate the effects of GLP-1 receptor agonists in the treatment of early PD.
These anti-inflammatory therapeutic approaches could be combined with existing therapies and it is likely that anti-inflammatory therapy might be more successful during early stages. Precision medicine in PD or other synucleinopathies aims to tailor medical treatment to the individual needs of each patient and it should consider factors such as genetic makeup, environmental exposure to risk factors, and lifestyle.308,323, 308,323 We should therefore consider any prior but also future exposure to infectious or inflammatory pathogens as it is an integral part of the patients’ environment and lifestyle. By identifying individuals who may benefit from anti-inflammatory treatments based on their genetic predispositions other tailored anti-inflammatory therapies can then be advised to address their specificneeds.
Implications for research
If infections are indeed involved in the etiology of synucleinopathies, it will necessitate research into the pathogens or type of infections that can contribute to the development of pathology. This will involve studying potential mechanisms by which infections can trigger or exacerbate neurodegeneration. Some of these mechanisms might overlap with the mechanisms involved in toxins, pesticides or other inflammatory molecules that could trigger pathology via environmental exposure.
We would also need to address methodological challenges. The lack of molecular diagnosis tools, the long prodromal phases and the clinical heterogeneity make it especially challenging to evaluate the effect of infections on the disease process and to establish a causal effect in the etiology of synucleinopathies. Not only the type of infection but also the timing of infection might be crucial for when pathology might or might not develop. New animal models that investigate how acute or chronic inflammation caused by infections could shed new light on how these diseases might arise.
It has been proposed that some of PD-linked pathogenic mutations with high prevalence and ancient origin (e.g., LRRK2, PINK, GBA and SNCA) might have provided a survival advantage when life expectancy was shorter and when infections were more common.296,300,305, 296,300,305 Impaired lysosomal function might influence susceptibility to neurodegenerative processes but could potentially have modified immune responses favorably against certain pathogens. Some studies have also suggested that overexpression of αSyn can restrict viral replication or viral assembly. 324 Now, with improved hygiene, and better anti-microbial treatments, these effects are less beneficial and exposure to virulent pathogens could instead lead to age-related neurodegenerative diseases as the same genetic risk factors will act as a disease facilitator at a later age. 280
The notion that these genes provided advantages in the face of infectious diseases is thus supported by their roles in the immune system. This is also seen in other neuroinflammatory diseases, like multiple sclerosis, where age-related and disease associated genetic variants were positively selected because they offered an evolutionarily advantage during times when infections were more common. 325 However, more detailed epidemiological and genetic studies are needed to substantiate these hypotheses and theories. This includes studying how these genetic variants might have been selected in populations when faced with high burdens of infectious diseases. Understanding these mechanisms could provide insights into why these genes are preserved in the human genome despite their association with disease in today’s context. Understanding these risk genes will help us to understand their physiological role and the molecular mechanisms in which they are involved during infection or inflammation. It could identify new potential therapeutic targets or help to devise new strategies for the prevention or treatment of high-risk populations. Importantly, it also warrants the use of novel therapies aimed at these pathways, as inhibiting the cellular pathways important for the immune response could potentially increase the susceptibility towards certain infectious pathogens in a population that is already moreimmunosenescent.
CONCLUSION
The hypothesis that infections might play a role in the etiology of synucleinopathies provides a framework for future research. The involvement of genetic risk factors in synucleinopathies, particularly those that regulate the immune response, underscores the potential interplay between genetic predispositions and environmental inflammatory triggers. Advancements in epidemiological methods have begun to shed light on specific pathogens that may be implicated in the development of these neurodegenerative diseases. By integrating genetic, immunological, and epidemiological insights, a multidisciplinary approach is required to further explore the origins and progression of synucleinopathies. Acknowledging the potential role of infections not only enriches our understanding of how synucleinopathies might arise but it also paves the way for the development of novel diagnostic tools and targeted treatments. By combining these approaches, preventive or precision medicine might hold potential to improve early diagnosis and treatment, to potentially delay disease onset and offer effective personalized care.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
W.P. has received commercial support for research from Myrtelle Gene Therapeutics. W.P. has received support as a consultant from Roche.