
Abstract
The discovery of a pathogenic variant in the alpha-synuclein (SNCA) gene in the Contursi kindred in 1997 indisputably confirmed a genetic cause in a subset of Parkinson’s disease (PD) patients. Currently, pathogenic variants in one of the seven established PD genes or the strongest known risk factor gene, GBA1, are identified in ∼15% of PD patients unselected for age at onset and family history. In this Debate article, we highlight multiple avenues of research that suggest an important - and in some cases even predominant - role for genetics in PD aetiology, including familial clustering, high rates of monogenic PD in selected populations, and complete penetrance with certain forms. At first sight, the steep increase in PD prevalence exceeding that of other neurodegenerative diseases may argue against a predominant genetic etiology. Notably, the principal genetic contribution in PD is conferred by pathogenic variants in LRRK2 and GBA1 and, in both cases, characterized by an overall late age of onset and age-related penetrance. In addition, polygenic risk plays a considerable role in PD. However, it is likely that, in the majority of PD patients, a complex interplay of aging, genetic, environmental, and epigenetic factors leads to disease development.
Keywords
INTRODUCTION
Parkinson’s disease (PD) is a clinically heterogeneous disorder [1, 2], with multiple genetic causes or contributors as well as environmental risk and protective factors identified [3, 4]. In most cases, it is likely that a complex interplay of a combination of genetic and environmental factors results in the disease and influences disease progression [5], and these factors may be different in different parts of the world [6] or exert differential effects depending on context [7, 8]. Here, we argue that the evidence supporting a genetic basis for PD is relatively more compelling than that for environmental factors.
EVIDENCE FOR AN IMPORTANT GENETIC ROLE IN PD
Many patients with PD have a positive family history
A substantial proportion of PD patients, perhaps up to ∼20%, have a positive family history [9]. Although it could be debated that this reflects not only genetic factors but also shared environmental exposure(s), some observations favor the former. Indeed, the discovery of SNCA, the first gene implicated in PD, was prompted when clinicians who were initially convinced that genetics played “no significant role in the etiology of PD” and “must be considered to be acquired” [10], encountered a very large kindred with an autosomal dominant pattern of PD transmission [11, 12]. Supportive evidence for the strong role of genetics in familial PD has been repeatedly demonstrated over the past three decades (e.g., references [13, 14], and in all regions of the world where this has been studied (http://www.mdsgene.org and [15]; Fig. 1). Recently, for example, in a study of Malaysian early-onset PD (EOPD), the rate of “solved” monogenic cases (i.e., where the cause of PD is attributed to pathogenic variant[s] in a single gene) increased from 21.7% overall to 48.5% when considering only the subgroup of EOPD patients with a positive family history [16]. Conversely, while it has been argued that the lack of a positive family history in the majority of PD patients points to environmental causation, many such individuals have, in fact, been shown to harbor a pathogenic genetic variant (e.g., 53.4% of the pathogenic variant-positive Malaysian EOPD patients had no history of PD in either the immediate or extended family [16]).
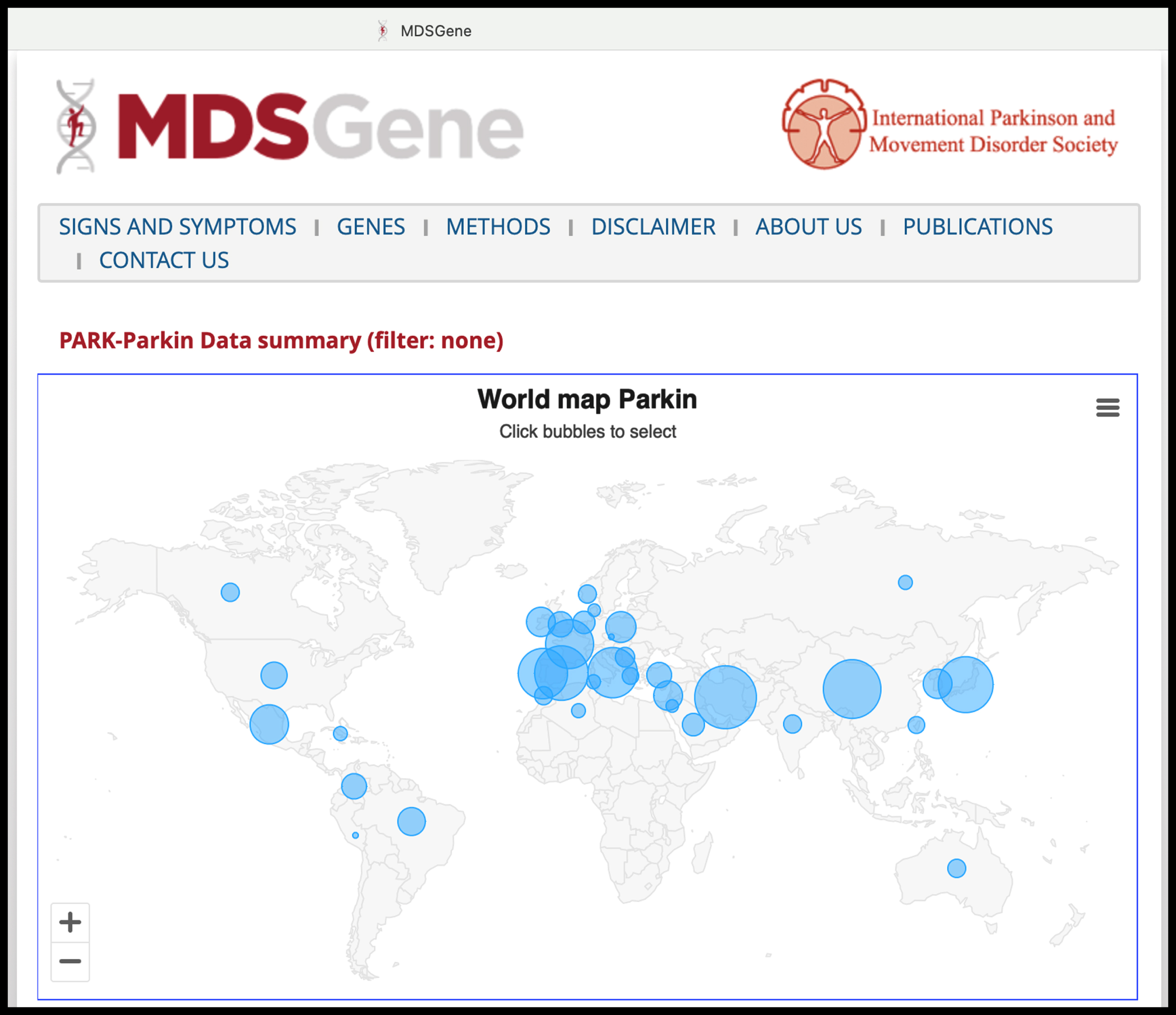
Screenshot from the MDSGene website (https://www.mdsgene.org, accessed on 5 November 2023), here depicting, as an example, the updated statistics on PARK-PRKN cases reported worldwide with individual-level data (almost 1,500 cases). The website also provides curated data regarding patients’ clinical characteristics and the pathogenicity classification of genetic variants.
A positive family history of neurodegeneration in related parkinsonian conditions such as progressive supranuclear palsy and multiple system atrophy, which have overlapping clinical features, pathology, and molecular mechanisms with PD [17–21], is reported in up to 20.4–33.0% [18, 22–24] and 40% [25] of cases, respectively, with genetic factors sometimes implicated [17, 26]. In contrast, environmental factors are still very poorly defined in these conditions [20, 28].
Pathogenic variants in some PD genes are highly penetrant for the disease
There are several established PD genes that cause monogenic/Mendelian forms of PD: SNCA, LRRK2, VPS35, and RAB32, which cause autosomal dominant PD, and PRKN, PINK1, and PARK7/DJ-1, which cause autosomal recessive PD [13–15, 29]. In some cases, pathogenic variant(s) in a single gene (e.g., whole-gene triplication of SNCA, or homozygous or compound heterozygous pathogenic missense and/or copy number variants in PRKN) is sufficient by itself to cause PD (i.e., demonstrating full penetrance) [12–15]. Remarkably, in some populations, these monogenic forms may even account for the majority of PD patients (e.g., >50% of PD patients attending a tertiary-care neurology clinic in the Malaysian state of Sabah are EOPD, of whom >50% have homozygous or compound-heterozygous PRKN exon deletions) [30]. Very high rates (sometimes exceeding 40%) of monogenic PD, or involving the strongest known risk factor gene GBA1, are also seen in selected populations such as the North African Arab-Berbers, Ashkenazi Jews, and Spanish Basques, especially involving LRRK2 and GBA1 [31–34], which have more variable (and age-related) penetrance and overall late age of onset [15].
We acknowledge, however, that the prevalence of monogenic PD in specialty clinics may not be generalizable to the population at large [35], and further studies are needed to accurately estimate the true prevalence of monogenic PD globally. On the other hand, the frequently quoted overall rates of monogenic PD (ranging from 2–3% [13], to 5–10% [36–38] or 15% [39]) may continue to increase due to new discoveries and broadening application of genetic technologies [40, 41], with inclusion of under-represented populations [42, 43]. The latter has recently been greatly bolstered by regional [44–46] and global collaborations such as the Global Parkinson’s Genetics Program (GP2; Fig. 2) [47–50]. It is worth noting here that in China and India (by far the two most populous countries in the world, being home to ∼2.9 billion people, and also with extensive diasporas globally [16]), the vast majority of PD patients with family pedigrees compatible with autosomal dominant inheritance remain “unsolved” (e.g., 95% of the 242 probands studied by Zhao et al. [37], and 100% of 44 probands in the study by Punia et al. which tested specifically for pathogenic LRRK2 variants [51]), suggesting that additional genetic determinants of PD remain to be discovered in these large populations [52, 53].
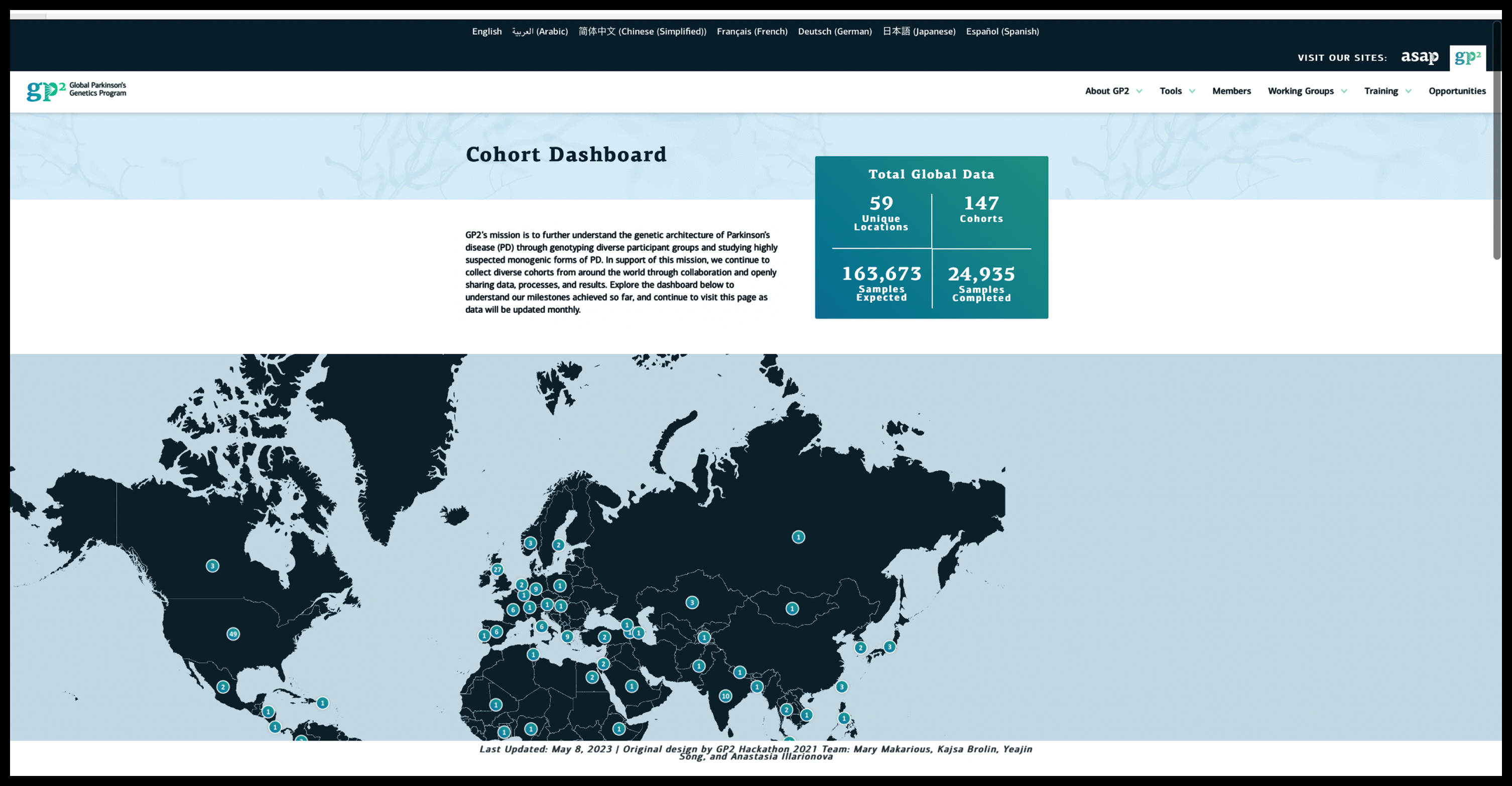
Screenshot from the Global Parkinson’s Genetics Program (GP2) website (https://gp2.org, accessed on 5 November 2023), depicting the updated statistics on an ever-growing number of contributing sites and samples.
Is there clustering of PD from environmental causes?
Conversely to the situation with genetics, to our knowledge, there have been extremely few reports of tight clustering of PD occurrence that have been convincingly demonstrated to be caused by environmental factors [54, 55]. Even for what is, to date, the most persuasive evidence for geographical linkage with PD (in Camp Lejeune, a military base in the USA), attributed to water contaminated with the solvent trichloroethylene (TCE) and other volatile organic compounds, the prevalence of PD was still “only” 0.33% (279 out of 84,824 exposed individuals; vs. 0.21% in veterans from a comparable but uncontaminated site) [28]. TCE has widespread and myriad uses, including clothes dry-cleaning [56], but to our knowledge, there have been no reports of clustering of PD among laundry or textile workers [57], other than an early study nearly 20 years ago involving three individuals working together in the office of a garment-manufacturing factory, one of whom also had a father with PD [58]. Thus, there is no environmental factor that is fully (or anywhere close to being fully) “penetrant” for PD. The highly toxic compound MPTP, which is the prototypical toxic cause for parkinsonism, was associated with seven human cases of parkinsonism [59], out of an estimated >400 (i.e., <2%) individuals thought to have been exposed to the synthetic heroin contaminant [60]. Moreover, it does not cause PD as we know it (in which disease processes take place gradually over many years, if not decades [3]), but rather results in a once-off “hit” with a severe but relatively selective nigral injury that is mostly non-progressive [61].
The study of cohabiting couples can identify environmental risk factors for disease because such couples are usually not genetically related but share residential exposures over a prolonged period. However, instances of conjugal PD appear to be very uncommon [62] and are usually considered to be coincidental [63].
Taken together, the above observations suggest that environmental factors are neither sufficient, nor necessary, to induce PD, and therefore cannot be considered a “predominant” cause for the condition.
What about sporadic (so-called “idiopathic”) PD?
In addition to pathogenic variants with high penetrance in PD genes, genomic DNA variation is now well established to contribute substantially to the risk of sporadic PD. To date, genome-wide association studies (GWASs) have identified >100 risk signals associated with PD at the population level [50, 64–67]. These common variants individually have relatively small effect sizes but in combination, can be associated with substantially elevated PD risk (e.g., with disease odds ratios [OR] of 3.4–6.1 among those with the highest decile of polygenic risk scores [PRSs] compared with the lowest-risk decile, in recent studies [64, 69]). Outside the PD field, it has been suggested that PRSs for common diseases such as coronary artery disease and type 2 diabetes, utilizing much larger sample sizes and improved algorithms, can identify individuals with risk equivalent to monogenic mutations [70] (although others have argued that the true effect size is likely to be much more modest [71–74]). Recently, research on genetic resilience factors that mitigate the effects of risk/pathogenic loci and reduce the susceptibility to PD is also emerging from GWA analyses [75].
Currently, the identified risk loci explain an estimated 16–36% of the heritable risk of PD [64], and efforts are ongoing to very substantially ramp up the recruitment of patients (to reach 200,000, including patients from diverse populations) [47–50], which will further advance understanding of the genetic determinants of PD on a global scale [13–15, 65].
Environmental factors act on a background of genetic vulnerability
Even for environmental exposures with an “established” role in PD etiopathogenesis, such as pesticide exposure, caffeine intake, and smoking, their downstream effects are likely to be mediated in part or strongly influenced by genetic factors [5, 8]. We list several examples here: Intriguingly, there is a suggestion in the literature that the lack of caffeine intake, which overall is associated with a ∼2-fold increased risk of developing PD, may be especially detrimental in patients with certain genetic forms of PD, such as those harboring the common Asian LRRK2 risk variants p.G2385R or p.R1628P (present in ∼5–10% of Asian PD patients [6, 76–78]), where odds ratios for PD were 8.6 and 4.6, respectively [79]. Findings consistent with caffeine’s greater association with resistance to genetic forms of PD were also reported among an international cohort of LRRK2-PD patients, most of whom harbored the p.G2019S LRRK2 variant seen in whites and North Africans [80]. A study exploring the neuroprotective effect of tobacco (containing nicotine) found that this may be mediated in part by SIRT6, which may have a pathogenic and pro-inflammatory role in PD and whose expression is strongly influenced by several SIRT6 single nucleotide polymorphisms (SNPs) [81]. Another study of the smoking effect on PD risk found that this varied by SV2 C genotype, ranging from being highly protective to neutral to even being harmful [45, 82]. Finally, a study found that the odds for developing PD from paraquat use was 1.5 in the presence of functional GSTT1 (encoding glutathione S-transferase T1 that provides cellular protection against oxidative stress) but increased to 11.1 with homozygous deletions of the gene which were present in a substantial proportion (22%) of patients [83].
Alterations in the gut microbiome (“dysbiosis”) have consistently been reported in PD patients over the past decade [84, 85]. While the microbiome is known to be significantly influenced by environmental factors such as diet and place of living [84, 86], genetic factors likely play key, if not essential, roles in determining if and how, various aspects of the gut-brain axis (e.g., gut dysbiosis or inflammation [87]) influence disease risk. For example, apart from rotenone models [88, 89], currently the most convincing animal models of gut dysbiosis in PD involve an induced genetic aberration, causing synuclein overexpression, mitochondrial dysfunction, or LRRK2 overexpression, suggesting that an underlying genetic vulnerability is usually needed for gut dysbiosis to trigger disease expression [84, 90–92].
Besides causation/development of disease, genetic factors can also have a significant influence on the disease trajectory
An increasingly highlighted example of this is the overall more rapid deterioration of motoric and cognitive-behavioral function and poorer survival in patients harboring GBA1 variants [93–95]. Some GBA1-PD patients also appear to respond less favorably to treatment with deep brain stimulation (DBS) [94, 96–98]. The converse is true for PARK-PRKN, where, on the whole, patients continue to show dopa-responsiveness even in the long term, exhibit less cognitive decline, and respond relatively favorably to DBS [96, 100]. These genetic factors are increasingly taken into consideration in the clinical management of patients, e.g., in the selection or counseling of patients for DBS [101, 102], and bring the field one step closer to realizing a personalized precision medicine approach for people living with PD [1, 103–105]. In sporadic PD, besides their association with disease risk, PRSs have also shown predictive value in PD phenotype or clinical outcomes [14, 106], such as age at disease onset [107]; motor progression [108]; development of dyskinesias [109], impulse control disorders [110] or cognitive decline (e.g., with one recent study reporting a hazard ratio of 4.8 for progression to PD dementia with the RIMS2 progression locus [111]); and response to medical (pharmacogenomics) [112] and surgical therapies (“surgicogenomics”) [113, 114].
In contrast, the impact of environmental factors, such as exercise [115–118] or dietary patterns (e.g., caffeine or alcohol intake [84]) on disease progression is much less well defined. That said, much remains to be understood with respect to genotype-phenotype correlations, e.g., patients with the exact same point mutation in genes linked to monogenic PD [98, 119–122] can sometimes still exhibit very different clinical courses, suggesting the presence of modifiers (genetic, epigenetic, and/or environmental) [5]. Even here, however, emerging evidence suggests a possibly more significant modifying role for genetic over environmental factors [123].
Understanding of genetic causation is paving the way for targeted therapies in PD
Genetic factors (and their related pathways) involved in the causation and progression of PD are prime targets for biomarker development and disease modification studies [14, 125]. For example, pathogenic variants in GBA1 or SNCA are known to be strongly linked to alpha-synuclein pathology [39], and novel biomarker approaches that enable in vivo interrogation of PD pathology (e.g., alpha-synuclein seeding assays [126–129]) are being exploited in new paradigms for biological classification/stratification of PD which will maximize the likelihood of finding successful disease-modifying therapies (e.g., targeting alpha-synuclein [130]). This has been the case in oncology, where implementation of precision medicine and a focus on genetically defined subtypes of disease have seen remarkable success in developing new and effective therapeutics [124, 131]. Most recently, a new monogenic cause, a pathogenic variant in the RAB32 gene has been found and independently confirmed to cause PD in a dominant fashion [29]. Interestingly the RAB32 protein interacts with LRRK2, and it is likely that additional genetic causes of PD will be found using new sequencing technologies. Finally, there is also a growing appreciation of a convergence of mechanisms underlying monogenic and complex forms of PD, with several of the genes discussed earlier causing monogenic PD also implicated in GWASs (e.g., SNCA and LRRK2), and likely causing relatively subtle changes in protein expression [132–137]. Thus, future genetics-targeted therapies could also potentially be deployed in (the much larger group of) sporadic PD patients.
Conversely, it is currently difficult to envision a scenario where, for example, knowledge of a patient’s remote history of neurotoxicant exposure could be directly “actionable”. Nevertheless, experimental work prompted by findings from epidemiological studies has revealed many valuable mechanistic insights (e.g., MPTP and other neurotoxicants such as paraquat or TCE causing mitochondrial dysfunction [138, 139] and more recently even pathological activation of LRRK2 kinase activity [140]). New avenues continue to be explored to understand the molecular mechanisms of PD based on epidemiological observations, for example with the study of caffeine [79, 141] or nicotine [81, 142] and related metabolites.
Research on environmental and lifestyle factors is in need of technological advances and better tools
Scientific advances depend not only on new ideas and paradigm shifts but also, to a large extent, on technological advances that make these leaps possible [143]. It has to be acknowledged that a crucial reason for the preponderance of evidence linking genetic status with disease causation and progression of PD is that genetics/genomics are much more tractable/easily ascertained with currently available technologies [65, 144], whereas environmental exposures are still difficult to assess and measure [140, 145] and such studies may be just “scratching the surface” [146]. Because of a lack of biological markers of exposure [140], studies of environmental factors often rely on patient retrospective self-report, which are “noisy” and prone to biases and confounding (e.g., recall bias and reverse causation), especially in a condition like PD where there is usually a long lag time spanning one or more decades between the exposure of interest and PD diagnosis [28, 147]. (Conversely, reverse causation is not an issue with genetics [which are fixed at conception], and indeed, this has been utilized for epidemiological studies using, for example, Mendelian randomization approaches [148, 149]). (That said, there is mounting evidence that somatic genetic changes, which take place in every person during development and tissue maintenance, are relevant in PD and related neurodegenerative disorders and can sometimes alter clinical presentation, for example, the age at disease onset [150–152]).
Moreover, people are often unaware of toxicants they are exposed to, as seen in the Camp Lejeune experience [28]. Epidemiological studies sometimes also lack sufficient granularity to be able to hone down to the individual level. In the Camp Lejeune study, for instance, exposure to TCE was inferred based on camp location, but the investigators could not be certain that all residents were exposed to biologically meaningful levels of contaminants [28]. Another recent study suggesting an association between air pollution and PD risk was based on participants’ residential addresses at the district level [153]. This is also exemplified in the maps presented by Professors Dorsey and Bloem (Fig. 2 in [138]), depicting geographic areas of overlap across the United States in PD incidence vis-à-vis use of/exposure to paraquat, TCE and other chlorinated solvents, and particulate matter.
Emerging studies utilizing a more comprehensive investigation of the “exposome” (i.e., the sum of all exogenous and endogenous environmental influences on the human body over the lifespan) and its impact on PD risk and progression will no doubt be an important area for future progress [140, 155]. These will investigate environmental exposures in a more holistic way, looking at multiple factors rather than reductionist approaches studying the effect of only a single environmental factor or class of factors [140, 156]).
The elephant in the room: the “Parkinson’s pandemic”
It has been argued that the rapid increase in PD cases worldwide, since its first formal description by James Parkinson slightly over 200 years ago (“from six to six million”) [55], must be driven by environmental factors (given the relative stasis of innate biology over the same time frame), particularly industrialization and the widespread use of toxicants [55, 138]. While there is little doubt that PD is common and poses an increasingly heavy burden on healthcare systems worldwide [3, 157], the argument may be more multi-faceted than it appears at first glance. One major issue relates to the increasing longevity of world populations with a concomitant increase in age-related disorders. On the surface, this phenomenon should similarly affect conditions like Alzheimer’s disease (AD) as much as it affects PD incidence and prevalence [138]. Still, there may be important differences between these age-related neurodegenerative disorders. For example, late-onset monogenic forms are well recognized in PD (e.g., LRRK2-PD), with disease penetrance being highly dependent on age [7, 158]; however, to our knowledge, late-onset monogenic AD has only very rarely been described [159, 160].
Another crucial point is the improved diagnosis of PD over time [161–164]. In the absence of reliable in vivo diagnostic biomarkers [3, 129], PD remains a clinical diagnosis, which can often be challenging [161–163, 165]. Indeed, Professors Dorsey and Bloem highlighted that approximately half of PD cases detected during an extensive multicenter epidemiological survey conducted in China in the late 1990s had been undiagnosed prior to the study [138, 166] (with rates of non-diagnosis as high as 90+% in rural communities [167]). It has been suggested that China’s rapid increase in PD cases (more than doubling in age-adjusted PD prevalence between 1990 and 2016) is due to rapid industrialization [168, 169], but it is likely that accompanying improvements in economic status and healthcare literacy, access to better healthcare (with improved diagnosis [161–164], as well as survival [164], of PD patients), and scientific interest in PD [6, 171], have contributed substantially to the increased prevalence of PD. Advances in technology (e.g., multimodal biomarker testing for genetic variants, synucleinopathy and/or neurodegeneration [39, 130], and artificial intelligence/machine learning-based diagnostic algorithms [171–173]), applied widely and non-invasively, are likely to further increase the rates of diagnosis of PD, including people in prodromal or even pre-symptomatic stages of the disease [126, 174].
Further research on gene-environment interactions is needed
Ultimately, gene-environment interactions are probably crucial in causing most cases of common “complex” human diseases, including PD [175], and the factors may be additive or synergistic [83]. Gene-environment interaction studies are therefore critically needed [5, 140], including further research on how environmental exposures disrupt epigenetic regulators of gene expression (without changing the underlying DNA sequence) [175]. Currently, research on gene-environment interactions [5, 140] and epigenetics in PD [176–178] is still in its infancy. This will be an exciting area to follow in the coming years, and developments here will be crucial to developing predictive and preventive approaches [5].
Returning to the earlier discussion on the Parkinson’s pandemic, it is instructive to learn from other non-communicable diseases. In the case of obesity, for example, an even larger pandemic has been documented [179], with the global prevalence tripling over the past four decades, now affecting >600 million individuals [180]. There is no doubt that changes in dietary patterns, such as the wide availability of calorie-dense processed food and drinks and physical inactivity, have contributed to this phenomenon. However, for what is considered by many to be a prototypical “lifestyle”-related condition, the heritability for obesity is estimated to be ∼40–70% [180–182], with the brain (which controls hunger and systemic energy metabolism) harboring most of the gene products and pathways that have been linked to obesity in hundreds of genetics studies [182]. Analyses of genome-wide gene-by-environment interactions over the past decade have revealed important insights and informed understanding of disease pathophysiology, although they remain challenging and require sample sizes in the hundreds of thousands or more [180]. As a side note, revolutionary anti-obesity drugs have very recently come to market [182]; these had their beginnings in research on genes related to the glucagon pathway [183].
CONCLUSION
Although we have collated and presented here what we believe to be robust evidence, accumulated over the past 30 years, in support of a strong genetic basis for PD, at the end of the day, we also salute, unreservedly endorse, and stand in solidarity with the advocacy efforts of our esteemed colleagues Professors Dorsey and Bloem, and others [55, 140], to curtail the widespread use of toxicants, to help curb the global burden of PD, and also for a healthier future overall [184, 185]. We whole-heartedly concur with a quote they recently highlighted from Jerry Ensminger, one of the personnel who served at Camp Lejeune who developed PD in his retirement (and whose young daughter died of leukemia while he was serving there), that: “The benefit of the doubt should go to the people, not the chemical” [56].
Footnotes
ACKNOWLEDGMENTS
We would like to thank Andrew Singleton and Huw Morris for insightful discussions on the role of genetics in the “Parkinson’s disease pandemic”.
FUNDING
SYL has received research grant funding from the Ministry of Higher Education Malaysia Fundamental Research Grant Scheme (FRGS/1/2020/SKK0/UM/01/2); University of Malaya Parkinson’s Disease and Movement Disorders Research Program (PV035-2017); and the Global Parkinson’s Genetics Program (GP2).
CK has received research grant funding from the Global Parkinson’s Genetics Program (GP2). The GP2 is funded by the Aligning Science Across Parkinson’s (ASAP) initiative and implemented by The Michael J. Fox Foundation (MJFF) for Parkinson’s Research (https://gp2.org). CK is also supported by the DFG and MJFF (outside GP2).
CONFLICT OF INTEREST
SYL has received consultancy or lecturing honoraria from the GP2 (the GP2 is funded by the Aligning Science Across Parkinson’s (ASAP) initiative and implemented by The Michael J. Fox Foundation for Parkinson’s Research (https://gp2.org)); Lundbeck International Neuroscience Foundation (Neurotorium) Editorial Board; International Parkinson and Movement Disorder Society (MDS); Eisai; Lundbeck; and Medtronic.
CK is a medical advisor to Centogene and Retromer Therapeutics and received Speakers’ honoraria from Bial and Desitin.