
Abstract
Slowing or halting progression continues to be a major unmet medical need in Parkinson’s disease (PD). Numerous trials over the past decades have tested a broad range of interventions without ultimate success. There are many potential reasons for this failure and much debate has focused on the need to test ‘disease-modifying’ candidate drugs in the earliest stages of disease. While generally accepted as a rational approach, it is also associated with significant challenges around the selection of trial populations as well as trial outcomes and durations. From a health care perspective, intervening even earlier and before at-risk subjects have gone on to develop overt clinical disease is at the heart of preventive medicine. Recent attempts to develop a framework for a biological definition of PD are aiming to enable ‘preclinical’ and subtype-specific diagnostic approaches. The present review addresses past efforts towards disease-modification, including drug targets and reasons for failure, as well as novel targets that are currently being explored in disease-modification trials in early established PD. The new biological definitions of PD may offer new opportunities to intervene even earlier. We critically discuss the potential and challenges around planning ‘disease-prevention’ trials in subjects with biologically defined ‘preclinical’ or prodromal PD.
Keywords
INTRODUCTION
Modified from Mahlknecht et al. (2022) [10], as permitted under the applying Creative Commons Attribution 4.0 International License.
Parkinson’s disease (PD) is unique among the neurodegenerative diseases for the availability of highly effective symptomatic therapies. The clinical efficacy of levodopa and other dopaminergic drugs is striking and, in many cases, able to almost completely control the cardinal motor features of the disease [1–4]. However, none of the available drugs to treat the symptoms of PD are able to slow the underlying progression of the disease and the increase in overall disability. The latter is driven by a combination of motor and non-motor features that characterize advanced PD and include levodopa-related response fluctuations and drug-induced dyskinesias, the emergence of drug resistant motor symptoms like freezing of gait and falls, as well as a plethora of increasingly severe non-motor problems including cognitive decline, dysautonomia and sleep-wake dysregulation [5]. Despite symptomatic therapy 50% of PD patients have been shown to meet pre-defined disability milestones and 22% are functionally dependent after only 5 years of disease [6, 7]. After 10–15 years more than >50% have developed hallucinations and/or dementia and >40% require institutional care [8, 9].
Disease-modification defined by preventing or delaying the progression of disability beyond symptomatic treatment effects is, thus, generally accepted as a key unmet need in the treatment of PD. From a regulatory perspective evidence for ‘disease-modification’ requires demonstration of effects of an intervention not only on clinical progression but also on underlying pathophysiological disease mechanisms, although it has been argued that demonstration of delay or prevention of clinical decline should be the prime anchor of definitions of disease-modification [10, 11] (see Panel).
The history of disease-modification trials in PD now spans about three decades during which a large number of well-designed and often large studies have used a broad range of drugs targeting different pathways potentially or definitely involved in the molecular pathology of the disease [12, 13] (Table 1). With the exception of two phase 2 trials of GLP1 agonists [14, 15], all of these efforts have yielded negative or, as in the case of the ADAGIO trial of the MAO-B inhibitor rasagilin [16], inconclusive results. Here we review potential reasons why so far almost all drug trials aiming to show disease-modification have failed, with a focus on novel targets and emerging perspectives of intervening at the earliest stages of disease.
Completed disease-modification trials in Parkinson’s disease (non-exhaustive list)
*Superiority to placebo in the rate of change in the UPDRS score between weeks 12 and 36, superiority to delayed-start treatment in the change in the score between baseline and week 72, and noninferiority to delayed-start treatment in the rate of change in the score between weeks 48 and 72. DAT, dopamine transporter; GBA1, glucocerebrosidase gene; H&Y, Hoehn and Yahr stage; MDS, Movement Disorder Society; PD, Parkinson’s disease; UPDRS, Unified Parkinson’s disease rating scale; MAO-B, monoamine oxidase Type B.
DISEASE MODIFICATION TRIALS IN PD: WHICH PATIENTS TO TARGET WITH WHAT INTERVENTION?
Numerous articles over the past decade have reviewed obstacles to demonstrate ‘neuroprotection’ or ‘disease-modification’ and potential reasons for the fact that no trial has yet led to the approval of a disease-modifying drug for PD [10–13, 17–21]. These include limitations in the translatability of preclinical findings of neuroprotection into the human disease, lack of reliable biomarkers for target engagement in early phase clinical development, challenges around selecting the right dose in clinical trials, as well as multiple trial design issues (see Table 2).
Challenges for disease-modification trials in Parkinson’s disease
Foremost among the latter are the selection of the right target population in a stage of disease that seems most accessible to disease-modification as well as the challenge of achieving sufficient trial durations required to detect clinically meaningful effects on disease progression.
Target populations for disease-modification trials in PD: early vs. later stages
With very few exceptions all trials of potentially disease-modifying agents have been conducted in newly diagnosed patients with disease durations of less than 2 to 3 years since the time of diagnosis with or without symptomatic drug therapy who were free of functional impairments (Table 1). This type of population is commonly referred to as ‘early’ PD as opposed to patients on treatment with levodopa plus other agents who have developed motor complications and are classified as ‘advanced’ PD.
Selecting subjects with early untreated PD has been accepted as a plausible strategy to ensure that the underlying pathology has not progressed too far for the respective pharmacological agent to still exert meaningful effects and at the same time allow for clinical comparisons with a placebo arm that are not confounded by effects of symptomatic therapies. One major reason why this approach has failed most of the time could be related to the slow progression of the severity of motor as well as non-motor symptoms in early PD. This leads to sensitivity issues of the ‘gold-standard’ scales that have been developed to assess symptom severity as well as functional impact. Most trials have used a primary endpoint of worsening of motor symptoms as measured by the UPDRS or MDS-UPDRS, which, because of its slow decline in early untreated PD, may be not sufficiently sensitive to detect statistically significant differences between active and placebo arms in this type of populations. Recent examples are the passive anti-synuclein immunotherapy trials of the monoclonal antibodies prasinezumab and cinpanemab, which failed to meet their primary endpoints of significant differences in progression of combined MDS-UPDRS parts I, II, and III scores over one year to 18 months [22, 23]. Over follow-up periods of 1 to 2 years the motor examination section of the MDS-UPDRS (part III) seems to be more sensitive compared to the patient report-based ‘motor experiences of daily living’ section (part II). This has been observed in several of the trials listed in Table 1 including the Prasinezumab trial, where progression of part III but not of part II scores (or combined part I, II, and III scores) was reduced in the active arms as compared to placebo [22].
Recent trials have moved into target populations with more advanced disease and recruited treated subjects with disease durations of up to 3 years or even beyond (see Table 1). Two of these assessing the efficacy and safety of the GLP-1 agonists exenatide [14] and lixisenatide [15] have indeed been positive showing significant differences in favor of active drug on a primary endpoint of motor worsening over 12 months as assessed by the MDS-UPDRS part III (as opposed to part II).
Selecting target populations in more advanced PD stages also improves chances to demonstrate clinical meaningfulness of effects from a putative disease-modifying intervention by using primary outcomes like time to development of functional disability or the occurrence of disability milestones in the motor or non-motor domains [13], although time-to event endpoints might require longer trial durations beyond the 12 to 24 months horizon of most previous trials (Table 1). An ongoing immunotherapy trial of prasinezumab in patients on stable symptomatic medication with disease duration of up to 3 years is using a primary outcome of time to a 5-point worsening of the MDS-UPDRS part III (NCT04777331).
One concern around testing disease-modifying agents in more advanced PD is related to the risk of the underlying pathology being too far advanced for the intervention to still exert clinical effects in spite of positive target engagement.
Selecting the right pharmacological targets
Identifying critical drug target within the pathogenetic cascade driving PD progression is another major challenge and the path to disease modification in PD is flanked by numerous failures of translation from preclinical proof-of-concept to clinical efficacy. Not least through the advances made in understanding the genetic architecture of PD multiple novel targets for disease-modifying pharmacological interventions have emerged and many of these are currently addressed in ongoing drug development programs [12, 24] (Fig. 1 and Table 3).
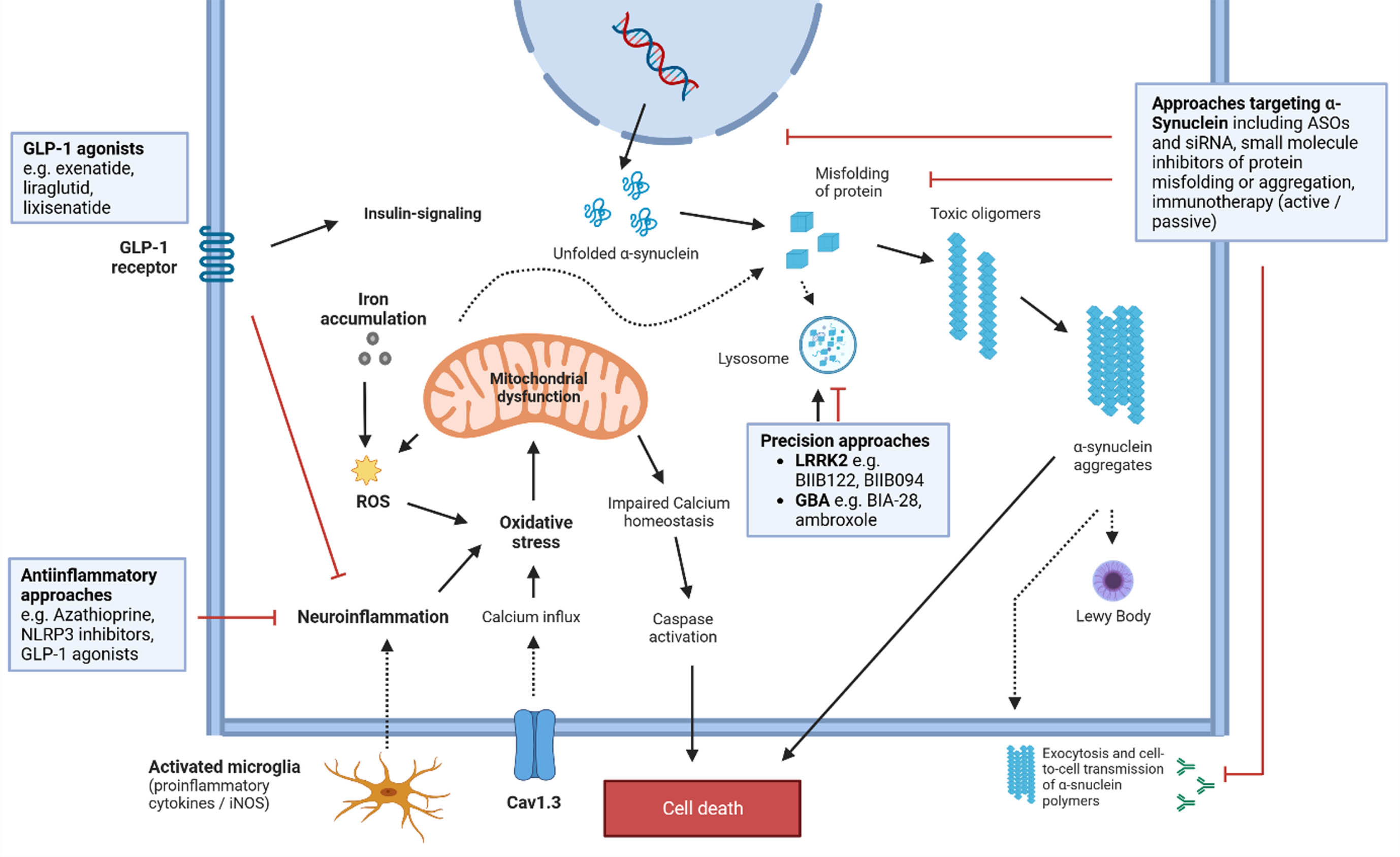
Mechanisms in the pathophysiological cascade of PD and potential treatment targets. See text and Table 3 for more details regarding drug candidates. ASO, antisense oligonucleotide; Cav1.3, Calcium channel, voltage-dependent; GBA, gene encoding for Glucocerebrosidase; GLP-1, Glucagon-like peptide-1; iNOS, Nitric oxide synthases; LRRK2, Leucine rich repeat kinase 2; ROS, reactive oxygen species; siRNA, small interfering RNAs.
Candidate drugs currently tested for disease-modification in PD (non-exhaustive list)
GCase, β-Glucocerebrosidase; FAF1, Fas-associated factor 1; HV, healthy volunteers; LRRK2, Leucine-rich repeat kinase 2; PD, Parkinson’s disease; PPAR γ, peroxisome proliferator-activated receptor; DAT, Dopamine transporter; GBA1, glucocerebrosidase gene; H&Y, Hoehn and Yahr stage; MDS, Movement Disorder Society; PD, Parkinson’s disease; UPDRS, Unified Parkinson’s disease rating scale; MAO-B, monoamine oxidase Type B.
Targeting specific pathogenetic pathways introduces the option of ‘personalized’ or ‘precision-medicine’ approaches to disease modification in PD, exemplified by recent drug trials targeting GCase or LRRK2 activity in PD populations harboring mutations in these genes [25]. The first large disease-modification trial phase 2 study in a genetic PD subtype enrolled 221 participants with one or more GBA1 gene variants to test if venglustat, a brain penetrant glucosylceramide synthase inhibitor, could slow the progression of combined MDS-UPDRS part II and III scores over 52 weeks [26]— following the rationale of ‘substrate-reduction’ that is a standard therapy for people with Gaucher’s disease and also been able to reduce α-synuclein pathology and behavioral deficits in rodent models. Patients were on dopaminergic therapy and had average disease durations of 4 to 5 years from time of diagnosis. The trial failed its primary endpoint and change from baseline to week 52 in combined MDS-UPDRS part II and II scorers was even numerically greater in the venglustat group, although there was target engagement with a 75% reduction of CSF glucosylceramide levels. While the negative results of this trial show that the principle of substrate-reduction for Gcase is unlikely to provide benefit in GBA-PD, it does not invalidate targeting Gcase activity directly by molecular chaperones. Several such agents are currently tested in phase 2 and 3 trials including ambroxol (NCT05778617, NCT05830396) and BIA-28 (NCT05819359).
LRRK2 inhibitors are another group of candidate drugs for a ‘personalized’ approach to disease-modification in PD. BIIB122 is an oral selective, brain penetrant inhibitor of LRRK2 for which safety and target engagement have been shown in both healthy volunteers and subjects with PD [27]. A phase 3 trial of this drug in PD patients carrying a pathogenic LRRK2 mutations was set up to measure effects on time to predefined worsening in the MDS-UPDRS II and III (NCT05418673) and a corresponding phase 2 trial is enrolling PD patients without mutation (NCT05348785).
DISEASE PREVENTION TRIALS IN PD: CHALLENGES AND FUTURE OPPORTUNITIES
Preventing or delaying ‘phenoconversion’ in subjects meeting criteria for ‘prodromal’ PD has been discussed as a new approach of ‘disease-modification’ trials that would conceptually become a type of ‘disease-prevention’ effort [10, 28].
Such target groups can now be defined by established multifactorial screening algorithms for prodromal PD such as the MDS criteria [29], although— despite their high specificity— their sensitivity and positive predictivity for early conversion in population-based cohorts seem suboptimal [30–32]. Other approaches to identify such individuals target single specific prodromal markers with subsequent enrichment steps like hyposmia followed by DAT-Scan as exemplified in the PARS study [33]. In idiopathic RBD, the strongest and most specific marker for PD and other α-synucleinopathies, conversion rates are only around 6% per year [34] and long-term series have reported median latencies from presumed RBD onset to clinically overt disease of 12–14 years and from diagnosis of idiopathic RBD to clinically overt disease of 6 years [35, 36]. These drawbacks of long delays to phenoconversion can potentially be overcome by enrichment strategies like adding further risk-markers such as hyposmia, subtle motor dysfunction, or subtle cognitive decline, which have all been shown to indicate higher conversion rates over shorter periods of time in subjects with idiopathic RBD [34, 37]. Despite these obstacles, prodromal cohorts are highly appealing as targets for disease-modification trials. On one hand the neurodegenerative process has already caused clinical symptoms that can be monitored for study purposes and that may also enhance motivation for individuals to participate in clinical trials. On the other hand, neurodegeneration may not have progressed too far to be modified by putatively neuroprotective interventions. All the approaches mentioned above in prodromal cohorts have yielded a maximum conversion rate of approximately 60% over 5 years or less [10, 34]. Before inclusion of participants into disease-modification trials it would be desirable to have an additional highly specific and ‘confirming’ biomarker to further enhance likelihood of a true prodromal state and to homogenize groups of prodromal individuals. Evidence of pathogenic α-synuclein on SAA would lend itself to such a purpose as it shows presence of the pathological hallmarks of synucleinopathies and it is highly specific for these disorders. Indeed, in line with the overall conversion rates in idopathic RBD of >80%, positive SAAs from CSF have been detected in around 90% in different series of idiopathic RBD patients [38–41].
Targeting asymptomatic or ‘preclinical’ disease stages could come even closer to an ultimate goal of ‘disease-prevention’ in the sense of completely preventing development of clinical disease in a subject’s lifetime [42]. Genetic PD subtypes are of particular interest in relation to disease-prevention trials for a number of reasons. First, these patients have defined molecular defects that are linked to the pathophysiology of their disease and thus allow for a precision-medicine approach to treatment [25]. In addition to reduced pathophysiological heterogeneity there is also less clinical heterogeneity in terms of disease progression in such cohorts [43]. Finally, asymptomatic individuals harboring mutations in PD genes are conceptualized as being in a ‘preclinical’ state of disease and intervening in this period could be more effective compared to later stages where pathology has progressed to override compensation and causes clinical symptoms [44]. Nonetheless, there are significant obstacles in the way of implementing disease-prevention trials in healthy carriers of PD-associated genes. These include the problem of recruitment of a trial population given that even GBA1 mutations, which represent the most common risk gene for PD, only affect around 5–10% of PD subjects globally and the overall carrier frequency of the G2019S mutation of the LRRK2 gene has been estimated at 0.5% [43, 45]. Reduced penetrance is another significant problem. As many as 70% of LRRK2 or 90% of GBA1 mutation carriers will never develop PD [43, 44] and for those who will the time to developing clinical symptoms is unknown. This would pose great difficulty in selecting meaningful outcome measures for trials in these types of target population and lead to long trial durations, which would also be true for the subsequent group of preclinical PD subjects.
Recent attempts to develop frameworks for a biological definition of PD may offer new opportunities to better define and enrich target populations for both disease-modification as well as ‘disease-prevention’ trials. Two recently published proposals both anchor the diagnosis of PD (subsumed under the term of ‘Neuronal Synuclein Disease’ (NSD) in one of them) on the presence of biomarkers independent of clinical symptoms. The proposed biological anchors rely on the demonstration of disease-specific alpha-synuclein pathology by seed amplification assays (SAAs) in the CSF, the presence of highly penetrant PD mutations, and evidence for dopaminergic neurodegeneration by molecular imaging [46, 47]. These approaches follow the example of biological diagnostic concepts initially put forward as the ‘ATN’ system for Alzheimer’s disease [48] and allow for a PD or ‘NSD’ diagnosis in the earliest stages of disease, when affected individuals have not yet developed any clinical symptoms or signs. Asymptomatic individuals with positive a-synuclein SAAs are classified as ‘Parkinson’s type synucleinopathy’ [46] or stage 1 NSD [47], while the appearance of ‘prodromal’ motor or non-motor signs with sufficient likelihood of being disease-related are classified as ‘stage 2’ in the NSD integrated staging system (NSD-ISS) proposed by Simuni and colleagues. Such biological diagnostic definitions would both allow for a more accurate diagnosis of ‘prodromal’ PD then is currently possible by using clinical features as the main anchors and would also offer an objectively measurable tool to detect preclinical disease. The latter, although conceptualized for a long time [49], has only recently become practically tangible –first by genetic markers and now through the availability of highly sensitive in-vivo assays to detect disease-specific α-synuclein pathology [50, 51]. A ‘biological’ definition of disease not only allows to detect preclinical disease but also has the potential to delineate pathogenetic subtypes and further reduce heterogeneity in future trial populations. The most attractive vision for the future use of these approaches relates to the prospect of implementing disease-prevention trials in populations of biomarker defined preclinical disease, i.e., people with ‘Parkinson’s type synucleinopathy’ [46] or ‘stage 1 NSD’ [47]. The recruitment base for such trials would presumably be much larger than is the case for trials selecting mutation carriers (see above). However, there are considerable challenges to be addressed when trying to screen for and select individuals in the population for such efforts.
OUTLOOK: WILL DISEASE-PREVENTION TRIALS IN PRECLINICAL PD BECOME FEASIBLE?
Using the newly proposed criteria for a biological disease definition there are two potential groups of preclinical subjects that could be recruited for disease-modification trials, those defined by harboring fully penetrant PD gene mutations or those with positive α-synuclein SAAs. Healthy subjects with such genetic variants are, however, very rare and recruitment for clinical trials would pose a major challenge. More common pathogenic mutations such as in the LRRK2 and GBA genes are associated with incomplete penetrance and much lower disease risk. Designing disease-prevention trials in healthy carriers of the latter group of mutations would require enrichment by additional molecular or imaging biomarkers for early conversion in order to arrive at sufficient endpoints in time frames that are feasible for trials and relevant for patients. Similar problems around recruitment and outcomes have to be addressed when targeting preclinical subjects that are defined by a positive result on α-synuclein SAAs (corresponding to stage 1 NSD [47] or ‘Parkinson’s type synucleinopathy’[46]). Even if reliable large scale SAA testing became feasible through non-invasive sampling via plasma- or serum-based assays [52, 53], the significance of a positive test in the population is currently uncertain. There are no studies dedicated to the assessment of SAA in the elderly community and prevalence of positive SAAs as well as the risk for subsequent development of α-synucleinopthies are unknown. In addition, identifying such individuals is not trivial even if the rate of SAA positivity in healthy control groups was around 5% to a maximum of 10% across the different hospital-based cohorts [54]. This seems to be in line with earlier studies finding incidental Lewy bodies and nigral neuronal loss in more than 10% of individuals free of PD above age 60 in histopathological population-based cohorts [55, 56]. Based on these numbers trial screen failure rates of up to 20%, α-synuclein SAA screening would have to be performed in more than 7000 subjects to arrive at a total sample size of 300 subjects for a disease prevention trial. In addition, lag times to clinical symptoms and/or to markers of neurodegeneration such as striatal dopaminergic loss in preclinical SAA positive individuals are expected to be very long. For these reasons alone SAAs testing would make more sense in preselected individuals with prodromal features like RBD or hyposmia (see above), who may have α-synuclein SAA positivity rates of around 80%. Once such individuals are identified, there is a need for additional biomarkers that indicate early progression or conversion to clinical disease. Imaging evidence for neurodegeneration such as DAT-SPECT has been proposed for such purposes [46]. Enrichment strategies will be essential to reduce numbers needed for disease-modification trials and to avoid employing unnecessary interventions that may be associated to psychological stress in individuals that may never go on to develop clinically relevant disease.
Selecting outcome measures for disease-modification trials in preclinical target populations would also mean entering largely unchartered territory. Time to ‘phenoconversion’ has been previously discussed in relation to disease-modification trials in prodromal PD but its use in biologically defined asymptomatic individuals poses even greater challenge in the latter compared to the former (see above). Alternative outcomes like on composite scores of motor, cognitive and other non-motor progression combined with evidence for progression of neurodegeneration from imaging or molecular markers could be viable and powerful alternatives [10, 57–61]. Achieving quantification of α-synuclein SAAs or the introduction of α-synuclein PET-imaging and establishing correlations with such quantitative measures with clinical progression might have an impact for outcome measures in disease-prevention and disease-modification trials alike, although it is yet unclear whether disease-modifying compounds would or should indeed have an influence on such measures or not.
Selecting agents to be tested also raises additional issues in preclinical cohorts. Testing interventions in people free of any clinical signs of disease and disability requires a level of safety that would have to practically exclude any risk of serious adversity. While this appears feasible for non-pharmacological approaches like lifestyle interventions, e.g., structured physical activity or nutritional programs [62], it is a very high bar for agents other than repurposed drugs with very well-established safety profiles and almost precludes trials of new and experimental interventions in preclinical individuals. The uncertainty if and when subjects meeting criteria for biomarker-based ‘preclinical’ disease will develop early clinical disease with functional impairment means that the burden of any ‘disease-preventing’ intervention should be minimal in order to be acceptable to patients and authorities alike [42].
Apart from safety risks of experimental therapies there are important additional ethical considerations to be addressed [63]. One of these concerns the phenomenon of ‘overdiagnosis’, which is a well-known problem for controversial screening tests and refers to the detection of subclinical disease (sometimes called pseudodisease), which would not have become manifest clinically in someone’s remaining life time (e.g., prostate cancer through prostate-specific antigen screening) [64]. ‘False-positive’ results of a screening procedure give rise to unnecessary further diagnostic tests and create worries of having a disease that may never manifest. Furthermore, depending on a given health care system, a positive screening result, e.g., a diagnosis of NSD, can have significant impact on the access to health care or life insurances [65]. Risk disclosure strategies should therefore take many factors into account, related to the screening test itself (e.g., its evidence, accuracy, reliability etc.) as well as to the subjects including education, social background, comorbidities, age, cognitive status, etc. Studies or trials in presymptomatic PD should therefore offer careful individualized counseling of participants and an option for psychosocial support.
Despite of all these challenges defining disease by the detection of underlying molecular pathology (‘biological definition’) marks a major step forward for efforts to achieve a reliable prodromal or even preclinical diagnosis which will eventually open the door to prevention also in the field of PD.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
P.M. reports lecture fees from AbbVie outside the submitted work. W.P. reports consultancy and lecture fees in relation to clinical drug development programmes for PD from AC Immune, Alterity, AbbVie, Affiris, BIAL, Biogen, Britannia, Lilly, Lundbeck, Merz, Neuroderm, Neurocrine, Roche, Sunovion, Stada, Takeda, UCB and Zambon, all outside the submitted work.