
Abstract
Background:
Various cellular pathways have been implicated in the transfer of disease-related proteins between cells, contributing to disease progression and neurodegeneration. However, the overall effects of protein transfer are still unclear.
Objective:
Here, we performed a systematic comparison of basic molecular mechanisms involved in the release of alpha-synuclein, Tau, and huntingtin, and evaluated functional effects upon internalization by receiving cells.
Methods:
Evaluation of protein release to the extracellular space in a free form and in extracellular vesicles using an optimized ultracentrifugation protocol. The extracellular effects of the proteins and extracellular vesicles in primary neuronal cultures were assessed using multi-channel electrophysiological recordings combined with a customized spike sorting framework.
Results:
We demonstrate cells differentially release free-forms of each protein to the extracellular space. Importantly, neuronal activity is distinctly modulated upon protein internalization in primary cortical cultures. In addition, these disease-related proteins also occur in extracellular vesicles, and are enriched in ectosomes. Internalization of ectosomes and exosomes by primary microglial or astrocytic cells elicits the production of pro-inflammatory cytokines, and modifies spontaneous electrical activity in neurons.
Objective:
Overall, our study demonstrates that released proteins can have detrimental effects for surrounding cells, and suggests protein release pathways may be exploited as therapeutic targets in different neurodegenerative diseases.
INTRODUCTION
Neurodegenerative diseases are associated with the progressive loss of a variety of brain functions due to the loss of different types of neuronal cells. Despite characteristic clinical manifestations, these disorders share common neuropathological features and cellular alterations, such as the aggregation and accumulation of disease-related proteins in relatively specific regions of the brain [1]. Alpha-synuclein (aSyn)-containing aggregates are typical in Parkinson’s disease (PD) and other synucleinopathies, hyperphosphorylated Tau-containing inclusions are typical in tauopathies, and mutant huntingtin (Htt)-containing inclusions are typical in Huntington’s disease [2–4].
The progressive accumulation of protein pathology in different brain regions [5–7] and the observation of aSyn Lewy body (LB) pathology fetal dopaminergic neurons grafted in the brains of PD patients led to the hypothesis that the progression of different neurodegenerative diseases may be correlated with the transfer of pathological proteins from sick cells to healthy cells [8, 9]. Consistently, injection of Tau aggregates into the brains of transgenic animals induces pathology along connected brain networks [10, 11]. Furthermore, a similar process was hypothesized to occur also in monogenic forms of neurodegenerative diseases after the observation of mutant Htt aggregates within fetal striatal allografts in the brains of Huntington’s disease patients [12].
The old brain is characterized by multi-morbidity, with the simultaneous accumulation of different types of protein pathology [13–15]. In addition, abundant Tau-related pathology can be observed in the brains of PD and Huntington’s disease patients, suggesting that multiple proteins may jointly contribute to the pathophysiology of different neurodegenerative disorders [16, 17]. Furthermore, the propagation of the pathological proteins between cells and across anatomical connected regions is consistent with the progression patterns described in different neurodegenerative diseases [18–20]. However, the precise molecular mechanisms underlying the spreading of pathology, and the relative contributions of each of them towards spreading, are still unclear. At a fundamental level, it is also unclear whether cells utilize similar pathways for releasing proteins, such as aSyn, Tau or Htt, and how the released proteins affect neighbouring cells.
Several conventional and unconventional pathways have been implicated in the cell-to-cell transfer of proteopathic seeds [20, 21]. The conventional secretory pathway requires the presence of a signal peptide sequence in the secreted protein that is then translocated to the endoplasmic-reticulum (ER), and sorted through Golgi-derived vesicles that, ultimately, fuse with the plasma membrane, thereby releasing their content/cargoes to the extracellular space [22]. Alternatively, cargoes can be sorted and released through unconventional pathways, resulting in the release of proteins in free forms, in extracellular vesicles (EVs) [23], or via tunnelling nanotubes, structures that enable the direct transfer of cargoes between connected cells [24].
EVs play various roles in the CNS, such as in intercellular communication, or the removal of toxic materials from the cell. In this context, they may also contribute to the transfer of pathogenic proteins in neurodegenerative diseases. EVs may be classified as exosomes or microvesicles (also known as ectosomes). They differ significantly in size, mechanism of biogenesis, and in protein, lipid, and nucleic acid content [25]. Exosomes (30–100 nm in diameter) originate from the multivesicular bodies (MVBs) and are released upon the fusion of MVB with the plasma membrane. In contrast, ectosomes (100–500 nm in diameter) are formed by the outward budding of the plasma membrane [26]. Recently, several studies reported that exosomes can contain proteins associated with neurodegenerative diseases and, therefore, may be explored as disease biomarkers [23, 27]. However, the role of ectosomes in the pathogenesis of neurodegenerative diseases, and the general effects of EVs in neuronal activity remain largely unknown [28].
More recently, another unconventional secretion mechanism known as misfolding-associated protein secretion pathway (MAPS), was described to export misfolded proteins [29]. This mechanism uses the ER-associated deubiquitylase USP19 to preferentially export misfolded cytosolic proteins through the recruitment of proteins to the ER surface for deubiquitylation. These cargoes are then encapsulated into late endosomes and secreted to the extracellular space [29]. After internalization, misfolded proteins may act as seeds to template the misfolding and aggregation of their physiological forms [30, 31].
Here, we developed stable cell lines expressing aSyn, Tau, and Htt exon 1 (carrying either 25 or 103 glutamines, 25QHtt and 103QHtt, respectively) fused to EGFP in order to afford a systematic comparison of the various proteins. Our results demonstrate that the different disease-related proteins are released, as free forms and in EVs, at different levels. Overall, 25QHtt-EGFP, aSyn-EGFP, and EGFP-Tau were found at higher levels in the cell media than 103QHtt-EGFP. We observed similar results when these proteins were expressed in primary cortical neurons or expressed without the EGFP tag, suggesting that the process of release to the extracellular space is mainly dependent on the protein properties. Furthermore, we modelled the occurrence of the proteins in the extracellular space, as when proteins are released from cells, and assessed their effect in the spontaneous firing activity and bursting events of mature primary cortical neurons using multi-electrode arrays (MEA). Monomeric 43QHtt induced discernible alterations in the bursting properties of the cells when compared with 23QHtt or with vehicle-treated cells, suggesting detrimental effects of the polyglutamine expansion on neuronal activity. Interestingly, Tau internalization resulted in increased neuronal activity, with cells exhibiting shorter bursts and higher intra-burst spike frequency, in contrast with only minor alterations observed with aSyn. These results suggest that intrinsic properties of aSyn, Tau, or Htt present in the extracellular space, and not necessarily the levels of the proteins, modulate their effect on neuronal activity.
Interestingly, aSyn, Tau, and Htt are present at higher levels in ectosomes than in exosomes, without altering the overall proteome of the vesicles. Additionally, the internalization of EVs by microglial or astrocytic cells elicited an increase in the levels of IL-6, IL-1β#x03B2;, and TNFα#x03B1; pro-inflammatory cytokines. Microglial cells also displayed an increase in p62 and LC3 puncta, suggesting the activation of autophagy for digesting the EVs. Finally, neuronal cells also internalized ectosomes and exosomes enriched in aSyn, Tau, or Htt and, consequently, exhibited cell bursting irregularities that, overall, correlated with the type of EV used.
Our results indicate that common cellular mechanisms may be used for the transfer of aSyn, Tau, and Htt between different cell types. Interestingly, we report that these proteins are handled differently depending on the receptor cell. We posit that the identified similarities and differences between the release and extracellular effects of the three proteins suggest the need for careful consideration of possible targets for therapeutic intervention in different diseases.
MATERIALS AND METHODS
Ethics statement
Mice (C57BL6/J#00245 background) were bred and maintained under specific pathogen free conditions in the animal facility of the University Medical Center Göttingen (Göttingen, Germany). All animal procedures were performed in accordance with the European Community (Directive 2010/63/EU), institutional and national guidelines (license numbers 19.3213 and T 20.7).
Primary cultures
Neuronal cultures
C57BL6/J#00245 wild-type E15.5 embryos from the animal facility of the University Medical Center Göttingen were used for the preparation of primary cortical neuronal cultures, as previously described [28]. In detail, pregnant animals were anesthetized using carbon dioxide intoxication and the embryos extracted from the uterus. After removal of the meninges, cortex was dissected under a stereomicroscope and the tissue was transferred to ice-cold 1x Hanks’ balanced salt solution (CaCl2 and MgCl2 free) (HBSS; Gibco Invitrogen, CA, USA) supplemented with 0.5% sodium bicarbonate solution (Sigma-Aldrich, MO, USA). Trypsinization was performed at 37°C for 15 min (100μ#x03BC;L of 0.25% trypsin; Gibco Invitrogen), and the reaction was stopped with the addition of 100μ#x03BC;L fetal bovine serum (FBS; Anprotec, Bruckberg, Germany) and 100μ#x03BC;L DNase I (0.5 mg/mL; Roche, Basel, Switzerland). After dissociation, the cell suspension was centrifuged at 300xg for 5 min and then resuspended in pre-warmed neurobasal medium (Gibco Invitrogen) supplemented with 2% B27 supplement (Gibco Invitrogen), 0.25% GlutaMAX (Gibco Invitrogen) and 1% penicillin-streptomycin (PAN Biotech, Aidenbach, Germany). Cells were seeded on coverslips coated with poly-L-ornithine (0.1 mg/mL in borate buffer) (PLO; Sigma-Aldrich) or culture plates (Corning, Merck, Darmstadt, Germany) for immunocytochemistry and western blot experiments. Cells were maintained at 37°C with 5% CO2, and fresh medium was added every three days.
Microglial cultures
Primary microglia were obtained from mixed glial cell cultures from C57BL6/J#00245 wild-type P0 pups from the animal facility of the University Medical Center Göttingen, as previously described [32]. Briefly, meninges were removed from the isolated brains and the tissue was collected into ice cold 1x HBSS (Gibco Invitrogen). The supernatant was removed, the brains were washed three times with HBSS solution (without Ca2 +, Mg2 + and phenol) (PAN Biotech) and incubated with 0.05% trypsin-EDTA (PAN Biotech) in a water bath at 37°C during 10 min. Trypsin was aspirated and the digestion was stopped by adding 0.5 mg/mL DNase I (Roche) in microglia medium [Dulbecco’s modified Eagle’s medium (DMEM; PAN Biotech) supplemented with 0.5% penicillin-streptomycin (PAN Biotech) and 10% FBS (Anprotec)]. Tissue was shortly incubated for 2–3 min at 37°C in the water bath and carefully homogenized into single-cell suspensions with a glass pipette. Suspension was centrifuged for 10 min at 800xg, the supernatant was discarded, and the pellet was resuspended in microglia medium. Cell suspension was plated into T75 flask (Corning, Merck) and incubated overnight at 37°C and 5% CO2. On the following day, two days in vitro (DIV2), the cells were washed three times with pre-warmed HBSS solution (PAN Biotech), one time with microglia medium and incubated with new medium until the next day. On DIV3, cell medium was replaced once more. On DIV5, the culture was stimulated by substitution of one third of the medium with L929 medium previously prepared in aliquots (see description in the cell lines section). The first harvest was performed on DIV8 by mild shaking and collection of the floating microglial cells. Medium was then centrifuged for 10 min at 800xg, the pellet resuspended in microglia medium, and cells were plated into culture plates previously coated with PLO (0.1 mg/mL in borate buffer) (Sigma-Aldrich) for immunocytochemistry, western blot, and gene expression studies. The culture was re-stimulated with new medium containing one third of L929 medium and incubated during three days until the next harvest. Three harvests were performed for the same culture.
Astrocytic cultures
Primary astrocytic cultures were prepared from C57BL6/J#00245 wild-type cerebral cortices of P0 pups from the animal facility of the University Medical Center Göttingen, as previously described [33]. After decapitation, meninges were removed, and the cortex was isolated from the brains and kept in ice cold 1x HBSS (Gibco Invitrogen). Tissue was digested in a fresh prepared solution of 0.25% trypsin (Gibco Invitrogen), 0.5 mg/mL DNase I (Roche), 1 mM EDTA (Carl Roth, Karlsruhe, Germany), 10 mM HEPES (Gibco Invitrogen), 2 mg/mL bovine serum albumin (BSA) (Sigma-Aldrich) in 1x HBSS (Gibco Invitrogen) at 37°C for 20 min. The reaction was stopped by adding astrocytes medium containing DMEM (PAN Biotech) supplemented with 10% FBS (Anprotec), 1% penicillin-streptomycin (PAN Biotech) and 25 mM HEPES (Gibco Invitrogen), followed by a short centrifugation at 800xg during 2 min. After aspiration of the supernatant, the tissue was dissociated with astrocytes medium until obtain a cell homogenate. After centrifugation at 800xg during 5 min, the pellet was resuspended in medium, and cells were plated in a T75 cm2 flask (Corning, Merck). Mixed culture was incubated at 37°C with 5% CO2 for three days. At DIV1 cells were washed with warm HBSS solution (PAN Biotech) and fresh media was added to the culture. The mixed astrocytic culture was agitated at DIV7 in an orbital plate shaker at 200 rpm for 40 min at 37°C. Cell culture agitation was repeated at DIV9 for 2.5 h at 250 rpm (37°C). A third shake was performed on DIV10 at 350 rpm, overnight at 37°C. After each agitation step, the supernatant was aspirated and the remaining cells in the flask were washed one time with warm HBSS solution (PAN Biotech) and fresh media was added to the cells. Cells were detached at DIV11 using 0.25% trypsin (Gibco Invitrogen) at 37°C for 5 min. Culture medium was added to stopped trypsin action, and the cell suspension was collected and centrifuged at 800xg for 5 min. Cell pellet was resuspended in medium and cells were seeded with fresh medium on plates or coverslips (Corning, Merck) coated with PLO (0.1 mg/mL in borate buffer) (Sigma-Aldrich) for immunocytochemistry, western blot, and gene expression studies at the appropriate density.
Cell lines
Mouse fibroblast L929 cells
Generation of L929-conditioned medium as performed as previously described [32]. L929 mouse fibroblast cells (kindly provided by Prof. Dr Hannelore Ehrenreich, MPI-EM, Göttingen, Germany) were plated into a T175 cm2 cell culture flask (Corning, Merck) with 100 mL culture medium for seven days containing DMEM (PAN Biotech) supplemented with 10% FBS (Anprotec) and 1% penicillin-streptomycin (PAN Biotech) at 37°C with 5% CO2. The media was then collected, sterilized by filtration with 0.22μ#x03BC;m filter (Sartorius, Göttingen, Germany) and stored at –20°C. The aliquots were freshly thawed when used for stimulation of the microglial cell culture.
Human embryonic kidney cells
Human embryonic kidney (HEK) 293 cells (ATTC, VA, USA) were maintained in DMEM medium (PAN Biotech) supplemented with 10% FBS (Anprotec) and 1% penicillin-streptomycin (PAN Biotech) at 37°C in a 5% CO2 atmosphere.
Transfection protocol
HEK 293 cells were seeded in cell culture plates (Corning, Merck) in the day before at the appropriate density. Transfection protocol was performed using Metafectene (Biotex, TX, USA) according to the manufacturer’s instructions and using equimolar amounts of the plasmids of interest. The following plasmids were used for the transfection protocol: pcDNA 3.1, pcDNA 3.1-aSyn, pcDNA 3.1-Tau (4R2 N), pcDNA 3.1-22QHtt exon 1 (1-90, CAG, ID CHDI-90000027, Coriell Institute), pcDNA 3.1-72QHtt exon 1 (1-90, CAG, ID CHDI-90001882-1, Coriell Institute), mCitrine-USP19 (Plasmid #78593, Addgene), mCitrine-USP19 C506 S (Plasmid #78594, Addgene). After 4 h of transfection, fresh medium was added to the cells (as described above in the cell lines section). Cells were collected or stained for further western blot or immunocytochemistry analysis after 24 h or 72 h as described in each experiment section. USP19 plasmids were kindly provided by Prof. Dr. Yihong Ye through Addgene [29], and Htt plasmids were kindly provided by NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research.
Lentivirus production
Production of FUGW-aSyn-EGFP, FUGW-EGFP-Tau, pRRL-CMV-25QHtt-EGFP-PRE-SIN, and pRRL-CMV-103QHtt-EGFP-PRE-SIN lentivirus was performed as previously described [34]. Htt lentiviral vectors were kindly provided by Prof. Dr. Flaviano Giorgini (Leicester University, Leicester, United Kingdom) [35]. HEK 293 cells were seeded in culture plates (Corning, Merck) and incubated in DMEM (PAN Biotech) supplemented with 10% FBS (Anprotec) and 1% penicillin-streptomycin (PAN Biotech) overnight at 37°C with 5% CO2. On the following day, cells were incubated with DMEM with 2% FBS (Anprotec), transfection medium for 5 h before transfection. Cells were transfected using calcium phosphate (CaPO4) precipitation method with a plasmid mix [57.9μ#x03BC;g vesicular stomatitis virus glycoprotein (VSVG) packing virus, 144μ#x03BC;g of Delta 8.9 packaging virus, and 160μ#x03BC;g of the plasmid of interest]. The DNA mix was added to 6 mL of 1x BBS (50 mM BES, 280 mM NaCl, 1.5 mM Na2HPO4) and 0.36 mL CaCl2 (2.5M CaCl2) was added to the mixture in a vortex shaker in the dark under sterile conditions. Before adding the mixture to the cells, solution was incubated 20 min in the dark. On the following day, cells were incubated with Panserin (PAN Biotech) supplemented with 1% of non-essential amino acids (MEM, Gibco Invitrogen) and 1% penicillin-streptomycin (PAN Biotech). Viruses were harvested 72 h after transfection and centrifuged at 3000xg for 15 min at 4°C. The supernatant was filtrated through a 0.45μ#x03BC;m filter (Sartorius) and incubated with 1x PEG solution (SBI System Bioscience, CA, USA) to pellet the viruses. Viruses were spin down by centrifugation at 1500xg during 30 min (4°C) after two days of incubation at 4°C. The pellet was resuspended in 100μ#x03BC;L Panserin (PAN Biotech).
Lentiviral infections
For neuronal cortical cultures, cells were infected at DIV14 with FUGW-aSyn-EGFP, FUGW-EGFP-Tau, pRRL-CMV-25QHtt-EGFP-PRE-SIN, and pRRL-CMV-103QHtt-EGFP-PRE-SIN and collected for further analyses at DIV19. Culturing conditions were the same as specified above (primary culture section).
Stable cell lines expressing aSyn, Tau, and Htt were established by lentiviral infection of HEK 293 cells with FUGW-aSyn-EGFP, FUGW-EGFP-Tau, pRRL-CMV-25QHtt-EGFP-PRE-SIN, and pRRL-CMV-103QHtt-EGFP-PRE-SIN. Cells were incubated during five days with the viruses and after three passages the infection rate was confirmed by microscopy (more than 90% of positive cells).
Lactate dehydrogenase assay
Cytotoxicity in the cell cultures (primary cortical neurons and cell lines) was assessed using the cytotoxicity lactate dehydrogenase (LDH) detection kit according to the manufacturer’s instructions (Roche). Furthermore, culture medium was centrifuged at 500xg for 5 min to pellet cell debris before used in the experiments.
Immunocytochemistry experiments
Primary cortical neurons and cell lines cultures after the treatments were washed with 1x PBS (PAN Biotech) and fixed with 4% of paraformaldehyde solution (PFA) for 20 min (home-made) at room temperature (RT). PFA autofluorescence was quenched by incubation with 50 mM of ammonium chloride (NH4Cl) solution for 30 min at RT. Cells were permeabilized with 0.1% Triton X-100 (Sigma-Aldrich) for 10 min at RT, and then incubated with 2% BSA in PBS (NZYTech) blocking solution for 1 h at RT. Incubation with primary antibody was performed overnight at 4°C [aSyn (1 : 1000, 610787, BD Biosciences, NJ, USA), aSyn (phospho S129) (1 : 500, ab51253, Abcam), α#x03B1;-tubulin (1 : 1000, 302217, Synaptic Systems), Glial Fibrillary Acidic Protein (GFAP) (1 : 1000, Z0334, Dako), Htt (1 : 500, MAB5374, Merck Millipore), Iba1 (1 : 1000, ab5076, Abcam), LC3 (1 : 500, PM036, MBL International), MAP2 (1 : 1000, ab11267, Abcam), Synaptophysin (1 : 500, 101002, Synaptic Systems), Tau (1 : 1000, MN1000, Thermo Fisher Scientific, MA, USA)]. Subsequently, the cells were washed with 1x PBS (PAN Biotech) and then incubated with fluorescence conjugated secondary antibodies for 2 h at RT [Alexa Fluor 488 donkey (1 : 1000, A21206, A21202, A11055, A21208, Invitrogen), Alexa Fluor 555 donkey (1 : 1000, A31572, A31570, A21432, Invitrogen), Alexa Fluor 633 goat (1 : 1000, A21050, Invitrogen), Alexa Fluor 633 donkey (1 : 1000, A21082, Invitrogen), and Alexa Fluor 680 donkey (1 : 1000, A10043, Invitrogen)]. Lastly, nuclei were counter-stained with DAPI (Carl Roth) and mounted with mowiol (home-made) for imaging experiments.
Western blots
Primary cortical neurons and cell lines cultures after the treatments were washed with 1x PBS (PAN Biotech) and lysed in RIPA buffer [50 mM Tris, pH 8.0, 0.15 M NaCl, 0.1% SDS, 1.0% NP-40, 0.5% Na-Deoxycholate, 2 mM EDTA, supplemented with protease and phosphatase inhibitors cocktail (cOmpleteTM protease inhibitor and PhosSTOPTM phosphatase inhibitor) (Roche)]. Lysate protein concentrations were determined using the Bradford protein assay (Bio-Rad, CA, USA). Samples containing 30μ#x03BC;g of protein were denaturated for 5 min at 95°C, loaded into 12% SDS-PAGE gels and transferred to nitrocellulose membranes using iBlot 2 (Invitrogen, CA, USA). Membranes were incubated in blocking solution containing 5% skim milk (Sigma-Aldrich) in tris-buffered saline (pH 8) with 0.05% tween 20 (TBS-T). Primary antibody incubation was performed overnight at 4°C diluted in 5% BSA (Sigma-Aldrich) in TBS-T. The following primary antibodies were used in this study: Alix (1 : 1000, ab117600, Abcam), aSyn (1 : 3000, 610787, BD Biosciences), aSyn (phospho S129) (1 : 1000, ab51253, Abcam), α#x03B1;-tubulin (1 : 1000, 302217, Synaptic Systems), β#x03B2;-Actin (1 : 10000, A5441, Sigma-Aldrich), Annexin-A2 (1 : 1000, ab178677, Abcam), APG5 L/ATG5 (1 : 1000, ab23722, Abcam), Flotilin-1 (1 : 1000, #18634, Cell Signaling), GFP (1 : 1000, 11814460001, Roche), Glial Fibrillary Acidic Protein (GFAP) (1 : 1000, Z0334, Dako), Huntingtin anti-N17 (1-8) [1 : 5000, kindly provided by Prof. Dr. Ray Truant (McMaster University, Ontario, Canada)], iNOS / NOS II (1 : 1000, 06-573, Upstate), Iba1 (1 : 1000, ab178846, Abcam), LC3 (1 : 1000, PM036, MBL International), MAP2 (1 : 1000, ab11267, Abcam), p62/SQSTM1 (1 : 1000, ab91526, Abcam), Synaptophysin (1 : 1000, 101002, Synaptic Systems), Tau (1 : 5000, A0024, Dako), USP19 (1 : 1000, A301-587A-M, Biomol). On the next day, the membranes were washed with TBS-T and incubated for 2 h with horseradish peroxidase (HRP) conjugated secondary antibodies [ECLtrademark Rabbit or Mouse IgG, HRP-linked whole antibody (1 : 10000, NA934 V or NXA931, Amersham)]. Subsequently, membranes were washed with TBS-T and incubated with a chemiluminescent HRP substrate (Millipore, MA, USA) for bands visualization in a chemiluminescence system (Fusion FX Vilber Lourmat, Vilber, France). Intensities of specific bands were normalized to a protein loading control or to the total protein levels marked using MemCodetrademark Reversible Protein (Thermo Fisher Scientific).
Native-PAGE
For native gel electrophoresis, samples were mixed with protein sample buffer (0.31 M Tris HCl pH 6.8, 50% Glycerol, 0.4% Bromophenol blue). Samples were loaded on pre-cast vertical Serva gels 4–16% (Serva, Heidelberg, Germany) and ran according to the manufacturer’s instructions using the appropriate buffers [50 mM BisTris-HCl pH 7.0 for the anode buffer; 50 mM Tricin, 15 mM BisTris-HCl pH 7.0 for the cathode buffer] (Serva). Proteins were transferred to nitrocellulose membranes using iBlot 2 (Invitrogen) and immunoblotting was performed as previously described in the western blot section.
Isolation of extracellular vesicles
Isolation of ectosomes and exosomes was performed as previously described [28]. HEK 293 cells expressing the proteins of interest were grown in conditioned medium (depleted of FBS-derived exosomes). Briefly, DMEM (PAN Biotech) supplemented with 20% FBS (Anprotec) and 2% penicillin-streptomycin (PAN Biotech) was centrifuged in polypropylene tubes (Optiseal; Beckman Coulter, CA, USA) in a fixed rotor (type 70 Ti, Beckman) during 16 h at 100000xg (4°C), as previously described [36]. The medium was subsequently diluted with DMEM medium (PAN Biotech) for a final concentration of 10% FBS (Anprotec) and 1% penicillin- streptomycin (PAN Biotech). Cells were seeded in T75 cm2 flasks (Corning, Merck) and incubated with fresh conditioned media during 24 h at 80% confluency. The media was collected, and protease and phosphatase inhibitors [(cOmpleteTM protease inhibitor and PhosSTOPTM phosphatase inhibitor) (Roche)] were added before centrifuging for 10 min at 300xg (4°C). A second centrifugation was performed at 2000xg for 20 min (4°C). Supernatant was transferred into ultra-clear tubes (Beckman Coulter) and ultracentrifuged in a swing rotor (TH-641 Sorvall; Thermo Fisher Scientific) during 90 min at 20000xg (4°C). Supernatant was carefully transferred into a new centrifuge tube and the pellet containing ectosomes was resuspended in ice cold PBS with protease and phosphatase inhibitors (PAN Biotech). The medium was centrifuged in a swing rotor (TH-641 Sorvall; Thermo Fisher Scientific) during 90 min at 100000xg (4°C) to purify exosomes. The pellet was resuspended in ice cold PBS with protease and phosphatase inhibitors (PAN Biotech). Ectosomal and exosomal pellets were diluted in PBS (PAN Biotech) and centrifuged at the correspondent velocities to wash and concentrate the vesicles fractions. The pellets were resuspended in 100μ#x03BC;l of ice cold 1x PBS with protease and phosphatase inhibitors (PAN Biotech). Protein concentrations were determined by the BCA assay following the manufacturer’s instructions (Thermo Fisher Scientific).
Labelling of extracellular vesicles
Extracellular vesicles were labelled with a fluorescent dye following a protocol previously described [28, 37]. Briefly, extracellular vesicles (60–100μ#x03BC;g of total protein in 50μ#x03BC;L PBS) were incubated with Alexa Fluor 633 C5-maleimide dye (200μ#x03BC;g/mL; Invitrogen) for 1 h in the dark at RT. Samples were resuspended in 10 mL PBS (PAN Biotech) and centrifugated during 90 min at 20000xg (4°C) for ectosomes and 100000xg for 90 min for exosomes (4°C) in a swing rotor (TH-641 Sorvall; Thermo Fisher Scientific). As a control, PBS was similarly incubated with the dye to confirm the removal of the dye excess.
NTA analysis
Extracellular vesicles size distribution and particle number were measured using nanoparticle tracking analysis (NTA) with NanoSight LM10 instrument (LM14 viewing unit equipped with a 532 nm laser) (NanoSight, Salisbury, UK). For each replicate, ectosomes and exosomes fractions were diluted in 1x PBS (PAN Biotech) to a final volume of 400 mL prior to analysis, according to the manufacturer’s recommendations (NanoSight). Measurements were recorded using NTA 2.3 software with a detection threshold of five, captured with a camera level of 16 at 25°C, in videos of 5 x 60 s repeated four times.
Electron microscopy
Electron microscopy (EM) images from extracellular vesicles was performed following a protocol previously described [28]. Isolated ectosomes and exosomes were bound to a glow discharged carbon foil covered grids. Samples were sained with 1% uranyl acetate (aq .) and then evaluated at RT using Talos L120 C transmission electron microscope (Thermo Fisher Scientific).
Immuno-EM
Immuno-EM images from HEK 293 cells expressing FUGW-aSyn-EGFP, FUGW-EGFP-Tau, pRRL-CMV-25QHtt-EGFP-PRE-SIN, and pRRL-CMV-103QHtt-EGFP-PRE-SIN were performed following a protocol previously described [38]. Briefly, cells were fixed with 4% formaldehyde and 0.2% glutaraldehyde in 0.1 M phosphate buffer. The cells were washed and then scraped from the dish in 0.1 M phosphate buffer containing 1% gelatin, spun down, and resuspended in 10% gelatin in 0.1 M phosphate buffer at 37°C. After spinning down again, the resulting pellets in gelatin were cooled on ice, removed from the tubes, and cut in small blocks. These blocks were infiltrated in 2.3 M sucrose in 0.1 M phosphate buffer, mounted onto aluminum pins for cryo-ultramicrotomy (Leica Microsystems, Vienna, Austria) and frozen in liquid nitrogen. Ultrathin cryosections were cut with a cryo-immuno diamond knife (Diatome, Biel, Switzerland) using a UC6 cryo-ultramicrotome (Leica Microsystems) and picked up in a 1 : 1 mixture of 2% methylcellulose and 2.3 M sucrose. For immuno-labelling, sections were incubated with 3H9 GFP primary antibody (#029762, ChromoTek, Planegg-Martinsried, Germany), followed by the secondary antibody (#112-4102, Rockland Immunochemicals, PA, USA) and then protein A-gold (10 nm) (Cell Microscopy Core, Utrecht, Netherlands). Sections were analyzed with a LEO EM912 Omega (Zeiss, Oberkochen, Germany) and digital micrographs were obtained with an on-axis 2048x2048-CCD camera (TRS).
Proteomic analyses of extracellular vesicles
For the proteomic analyses, ectosomes and exosomes samples were resuspended in Laemmli sample buffer and separated in SDS-PAGE gel, as previously described [28]. Briefly, the entire lane was cut in 23 gel pieces and tryptically digested [39]. Extracted peptides were analyzed in technical replicates by liquid chromatography coupled to mass spectrometry (LC-MS) on a Dionex UltiMate 3000 RSLCnano system (Thermo Fisher Scientific) connected to an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific). A 43 min gradient ranging from 8% to 37% acetonitrile on an in-house packed C18 column was used to separate the peptides (75μ#x03BC;m x 30 cm, Reprosil-Pur 120C18-AQ, 1.9μ#x03BC;m, Dr. Maisch GmbH, Ammerbuch, Germany) at 300 nl/min flow rate. MS1 spectra were acquired with 120000 resolution (full width at half maximum, FWHM) and a scan range from 350 to 1600 m/z. Within a cycle time of 3 s, precursor ions with a charge state between+2 and+7 were selected individually with a 1.6 m/z isolation window and were fragmented with a normalized collision energy of 35 by higher energy collisional dissociation (HCD). MS2 spectra were acquired in the ion trap with 20 % normalized AGC and dynamic injection time. Once selected precursors were excluded from another fragmentation event for 30 s. Raw acquisition files were subjected to database search with Maxquant (version 1.6.2.10) [40] against the reference proteome of Homo sapiens (downloaded on 02/19/2019) and the GFP fusion proteins. Default settings were used unless stated differently below. Fractions were defined according to the cutting of the gel lanes and experiments were defined on the level of technical replicates. Unique and razor peptides were used for label-free quantification except for the GFP fusion constructs where all peptides were used due to the difficulty of peptide assignment and protein grouping related to the EGFP tag. Label-free quantitative proteomics were performed with two unique peptides as a threshold to identify a protein in the sample.
All the proteomic data analysis were performed with Perseus (version and 1.6.15.0) [41]. The initial data was filtered for reverse hits, potential contaminants and hits only identified by site. Quantitative values were averaged across technical replicates ignoring missing values. A two-sample t-test was performed on biological replicates of the samples with an artificial within groups variance (s0) of 0.1 and a permutation-based FDR of 0.1 for multiple testing correction. These results were visualized in volcano plots.
Secretion and internalization assays
The collection of the cell media and evaluation of proteins secretion was performed using an adapted protocol from a previous study [42]. Briefly, 1 mL of medium from HEK cells expressing FUGW-aSyn-EGFP, FUGW-EGFP-Tau, pRRL-CMV-25QHtt-EGFP-PRE-SIN, or pRRL-CMV-103QHtt-EGFP-PRE-SIN was collected after 24 h and centrifuged for 5 min at 500xg at 4°C to pellet cell debris. Supernatants were concentrated ten times in an Amicon ultra 10K centrifugal filters (Millipore) following the manufacturer’s instructions. Only 30μ#x03BC;L of the 100μ#x03BC;L final volume was analyzed by western blot.
For the neuronal cell media, the DotBlot system was used to exclude the possibility that the proteins of interest could be retained in the centrifugal filters and change the results observed in the experiments. Briefly, 1 mL of the neuronal cell media was collected from primary cortical neurons expressing FUGW-aSyn-EGFP, FUGW-EGFP-Tau, pRRL-CMV-25QHtt-EGFP-PRE-SIN, or pRRL-CMV-103QHtt-EGFP-PRE-SIN and centrifuged for 5 min at 500xg (4°C). Only 300μ#x03BC;L of the 1 mL final volume was directly applied into the system in a nitrocellulose membrane (Bio-Rad).
For the internalization experiments, 1 mL of medium from HEK cells expressing FUGW-aSyn-EGFP, FUGW-EGFP-Tau, pRRL-CMV-25QHtt-EGFP-PRE-SIN, or pRRL-CMV-103QHtt-EGFP-PRE-SIN was collected after 24 h and centrifuged for 5 min at 500xg (4°C). Afterwards, the media was added to naüve HEK 293 cells for 72 h.
Flow cytometry experiments
As described in the previous section, naüve HEK 293 cells were incubated during 72 h with 1 mL of cell media from HEK cells expressing FUGW-aSyn-EGFP, FUGW-EGFP-Tau, pRRL-CMV-25QHtt-EGFP-PRE-SIN, or pRRL-CMV-103QHtt-EGFP-PRE-SIN. Subsequently, HEK 293 cells were washed with 1x PBS and trypsinized (PAN Biotech). Cell suspension was centrifuged during 5 min at 300xg (4°C). Cell pellet was resuspended in 1x PBS (PAN Biotech) to remove residual cell media and centrifuged for 5 min at 300xg (4°C). The supernatant was removed by aspiration and the cell pellet was resuspended in 1 mL of ice cold 1x PBS (PAN Biotech) with 0.1% of propidium iodide (Sigma-Aldrich). Cells without EGFP expression, treated with 0.1% triton-X (Sigma-Aldrich) or PBS alone were used as negative controls. Cells infected with EGFP lentivirus were used as positive control. 10 000 events were acquired on a FACSAria II flow cytometer (BD Biosciences). Flow cytometric data were analyzed with FlowJo Analysis Software (BD Biosciences).
Treatment of cells with extracellular vesicles
Primary cortical neurons and astrocytes were treated with 20μ#x03BC;g/mL of ectosomes or exosomes resuspended in 1x PBS (PAN Biotech) with protease and phosphatase inhibitors [(cOmpleteTM protease inhibitor and PhosSTOPTM phosphatase inhibitor) (Roche)]. Due to high toxicity, microglial cells were treated only with 10μ#x03BC;g/mL of extracellular vesicles. Treatment was performed in cortical neurons at DIV14, and cells were fixed or collected for further analyses at DIV15 (24 h treatment). Microglial and astrocytic cell cultures were treated in the day after their plating for 24 h, and then fixed or collected for further analyses. Pro-inflammatory stimulation was evaluated by the exposition to lipopolysaccharide (LPS; Thermo Fisher Scientific). Microglia cells were treated with 50 ng/mL and astrocytes with 100 ng/mL of LPS (Thermo Fisher Scientific). Culturing conditions were the same as specified above in the primary culture section.
Gene expression studies: RNA isolation and quantitative real-time PCR
Total RNA was extracted from microglial and astrocytic cultures 24 h after treatment using TRIzol Reagent according to the manufacturer’s instructions (Invitrogen). Reverse transcription of RNA to produce cDNA was performed using QuantiTect Reverse Transcription kit (Qiagen, MD, USA) following the protocol provided by the manufacturers. Quantitative real-time PCR (qPCR) analysis was performed on an Applied Biosystems Real-Time PCR Systems using SYBR Green Master Mix (Qiagen). The thermal cycler conditions were as following: 95°C for 10 min, then 40 cycles at 95°C for 15 s and 60°C for 25 s. Primers used in the quantitative real-time PCR experiments to evaluate the inflammatory markers were (mouse, sequence 5’ to 3’): IL-6 Forward- ATCCAGTTGCCTTCTTGGGACTGA, IL-6 Reverse- TAAGCCTCCGACTTGTGAAGTGGT, IL-10 Forward- GGTTGCCAAGCCTTATCGGA, IL-10 Reverse- CACTCTTCACCTGCTCCACT, IL-1β#x03B2; Forward- TCATTGTGGCTGTGGAGAAG, IL-1β#x03B2; Reverse - AGGCCACAGGTATTTTGTCG, TNFα#x03B1; Forward- CCCTCTCATCAGTTCTATGG, TNFα#x03B1; Reverse- GGAGTAGACAAGGTACAACC, β#x03B2;-actin Forward- GCGAGAAGATGACCCAGATC and β#x03B2;-actin Reverse- CCAGTGGTACGGCCAGAGG. Fold change expressions were calculated using the 2–ΔΔCT method, with β#x03B2;-actin as a reference gene [43]. Quantification in the graphs show the normalized relative quantity (NRQ) values compared with the control.
Treatment with recombinant monomeric protein
Primary cortical neurons were treated at DIV14 with recombinant monomeric protein until DIV19 (5 days). Final concentration of protein in the cultures was 100 nM and PBS was employed as negative control. Recombinant aSyn, full-length 2N4 R Tau, 23QHtt, and 43QHtt were prepared as previously described [44–46]. Culturing conditions were the same as specified above (primary culture section).
Multielectrode array experiments
MEA experiments were performed following standard protocols, as previously described [28, 47]. Primary cortical neuronal cultures were plated directly on 60MEA200/30iR-Ti-gr planar arrays (60 electrodes, 30μ#x03BC;m electrode diameter, 200μ#x03BC;m electrode spacing) (MultiChannel Systems, Reutlingen, Germany). The arrays were coated with poly-L-lysine overnight at 4°C (500μ#x03BC;g/mL in borate buffer; PLL) (Sigma-Aldrich) and the next day with laminin for 1 h at RT (5μ#x03BC;g/mL in distilled water) (Sigma-Aldrich). Neuronal cells were plated directly on top of the electrodes and treated with EVs or recombinant proteins. For the EVs recordings, cells were incubated with 20μ#x03BC;g/mL of ectosomes or exosomes containing the different disease-related proteins at DIV14 and recorded at DIV15, 24 h after the treatment. In the recombinant monomeric protein experiments, cells were incubated with 100 nM of aSyn, full Tau, 23QHtt exon 1 or 43QHtt exon 1 at DIV14 and recorded at DIV19, 5 days after the treatment. The neuronal activity was recorded using the MultiChannel MEA2100 system (MultiChannel Systems, Reutlingen, Germany) with temperature maintained at 35–37°C. Recordings started 10 min after translocation of the arrays from the incubator to the recording stage to avoid movement-induced artifacts, and the spontaneous activity was recorded for 5–10 min. The electrode signals were amplified, band-pass filtered (200 Hz to 3 kHz) and recorded digitally at 25 kHz, using the MultiChannel Experimenter software (version 2.17.7.0) (MultiChannel Systems, Reutlingen, Germany).
Spike sorting was carried out using a modified version of the Kilosort automatic sorting software, as previously described (available at: https://github.com/MouseLand/Kilosort [com/MouseLand/Kilosort] and https://github.com/dimokaramanlis/KiloSortMEA [dimokaramanlis/KiloSortMEA]) [28, 49]. Kilosort output was visually inspected and manually curated with the Phy2 software (https://github.com/cortex-lab/phy [cortex-lab/phy]). Only spike clusters (“units”) with a well-separated spike waveform and a clear refractory period were included in the data analysis and considered as originated from individual neuronal cells. The spike clusters were pre-processed and analyzed using custom-made MATLAB scripts (Version: 9.7.0, R2019b; Mathworks, MA, USA). The raster plots, voltage traces, average firing rate and spike amplitude were measured from the spontaneous activity of each recorded cell. In all the recordings, we observed the occurrence of frequent bursts [groups of spikes occurring rapidly and consecutively with short inter-spike intervals, less than few tens of milliseconds (ms)], followed by quiescent periods longer than normal inter-spike intervals [generally several seconds (s) in our recordings]. The burst activity usually occurred synchronously for multiple cells over the array and in our analysis, and we focused on this population-wide synchronized bursts for further data analysis. To detect the concurrent bursts, the population firing rate was computed as a histogram (100 ms bin size) of array-wide spiking activity. The peaks of the firing rate histogram were used to detect synchronous, array-wide bursts with at least 500 ms distance between two consecutive peaks. The peaks that were smaller than 1/5 of the largest peak were excluded as they do not correspond to array-wide synchronous activity. A time window of 650 ms around each peak (150 ms before to 500 ms after) was defined as the burst window (onset and offset of each burst). For each recorded cell, the spikes belonging to bursts were measured during the defined burst windows, and cells with fewer than six spikes across all their detected bursts were excluded from this analysis. From the detected bursts, the following parameters were calculated: 1) burst rate of the culture as the number of bursts per time over the duration of each recording; 2) inter-burst-interval as the time between the measured offset of a burst and the onset of the following burst, calculated for each pair of successive bursts in a recording; 3) burst duration for each cell as the time between the cell’s first and last spike during the burst window; 4) intra-burst-frequency as the rate of spikes occurring within a burst, averaged over all the detected bursts for each cell; and 5) percentage of spikes in bursts as the ratio of spikes occurring during bursts relative to the total number of spikes for each cell.
Confocal microscopy imaging
Imaging was performed on a Leica SP5 confocal laser scanning microscope equipped with hybrid detectors using Application Suite X software with 100x immersion objective lenses (Leica Biosystems, Wetzlar, Germany). Samples were excited using 405 Diode, argon and helium–neon 633 lasers, pinhole = 1, 0.2μ#x03BC;m thickness Z stacks and 2 averaging line-by-line. The acquisition settings were optimized to avoid underexposure and oversaturation effects and kept equal throughout image acquisition of the samples.
Quantifications and statistical analyses
Analyses of the images was performed using ImageJ software (National Institutes of Health) [50]. The EVs uptake in neuronal cells was measured by the ratio of the EVs signal area and cell area from different isolated areas chosen randomly within regions containing EVs signal. All data are presented as mean±SD. Data from at least three independent experiments and each replicate represents one independent experiment. To assess differences between two groups, two-tailed unpaired student t-test was performed using GraphPad Prism 9 software (GraphPad, CA, USA). To assess differences between more than two groups, significant differences were assessed by one-way ANOVA followed by multiple comparisons with significance between groups corrected by Bonferroni procedure using GraphPad Prism 9 software (GraphPad). Differences were considered to be significant for values of p < 0.05 and are expressed as mean±SD. For mass spectrometry, the spectral count differences between samples were considered to be significant for FDR values < 0.1 (see proteomic analyses section).
RESULTS
aSyn, Tau, and Htt are released to the extracellular space in a free form in different cell models
To evaluate the release of aSyn, Tau, and Htt to the extracellular space, we established stable HEK cell lines expressing the different disease-related proteins fused to EGFP (Fig. 1). We expressed two biologically relevant N-terminal exon 1 Htt fragments with either 25 or 103 polyglutamines (representing wild-type and mutant Htt, 25QHtt-EGFP and 103QHtt-EGFP, respectively) (Fig. 1A-B). While 25QHtt-EGFP, aSyn-EGFP, and EGFP-Tau expression was mainly diffused in the cytoplasm, 103QHtt-EGFP accumulated in inclusions in the nucleus and throughout the cell (Fig. 1A). Importantly, the use of the same cell type expressing the different proteins enabled us to directly compare the effect of the cellular machinery on protein release. We found that aSyn, Tau, and Htt were differentially released to the extracellular space (Fig. 1B). Cell lysates and conditioned media were assessed by SDS-PAGE, and protein levels were normalized to total protein levels using Memcode (Fig. 1C).
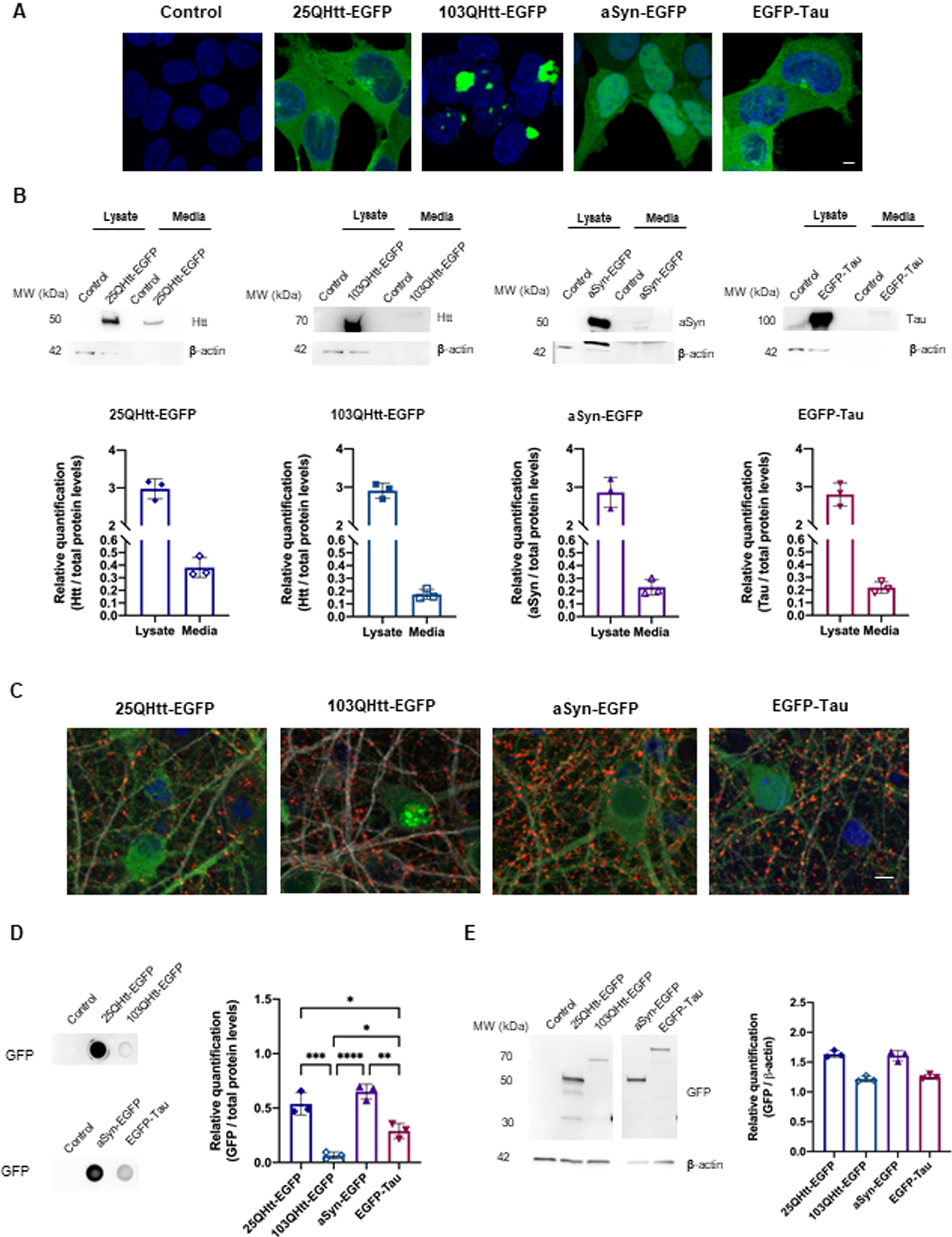
aSyn, tau, and Htt are released to the extracellular space. A) Representative images of HEK cells stably expressing 25QHtt-EGFP, 103QHtt-EGFP, aSyn-EGFP, or EGFP-Tau. Scale bar 5μ#x03BC;m. B) Immunoblots showing the protein levels in cell lysates and in the cell media of the different cell lines. Quantifications were normalized to total protein levels using MemCode (n = 3). Immunoblots were cropped for space purposes. C) Representative images of primary cortical neurons infected with lentiviral vectors encoding 25QHtt-EGFP, 103QHtt-EGFP, aSyn-EGFP, or EGFP-Tau, and immunostained for synaptophysin (red) and MAP2 (grey). Neurons were infected at DIV14 and cultured until DIV19. They were then fixed for imaging and the media was collected for further analyses. Scale bar 5μ#x03BC;m. D) Dot blot analyses of the protein levels released to the cell media of cells expressing the different proteins. Quantifications were normalized to total protein levels using Memcode (n = 3). Samples were applied in the same dot blot membrane and cropped for space purposes. E) Immunoblots showing the EGFP levels in whole-cell lysates. Quantifications were normalized to β#x03B2;-actin (n = 3). Immunoblots were cropped for space purposes. Significant differences were assessed by one-way ANOVA followed by multiple comparisons with significance between groups followed by Bonferroni correction. Differences were considered to be significant for values of p < 0.05 and are expressed as mean±SD, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
In addition, we evaluated the release of aSyn, Tau, and Htt in primary neuronal cultures (Fig. 1C–E). Cortical neurons were infected with lentiviral vectors encoding for the various proteins (at DIV14), to ensure homogeneous transduction (Fig. 1C). Cell media was collected at DIV19 and applied onto a dot blot system to assess the presence of the different proteins in the extracellular space (Fig. 1D). We observed that 25QHtt-EGFP was released at higher levels than 103QHtt-EGFP. Overall, 25QHtt-EGFP and aSyn-EGFP were released at higher levels compared with the other proteins (Fig. 1D), but these differences were not simply associated with the expression levels in the cells (Fig. 1E).
To rule out that secretion was associated with the presence of the EGFP tag, we expressed untagged aSyn, Tau, and Htt proteins in HEK cells (Supplementary Fig. 1A). Consistently with the previous data, we observed higher levels of 22QHtt, aSyn, and Tau in the cell media, and lower levels of 72QHtt (Supplementary Fig. 1B). Lactate dehydrogenase (LDH) cytotoxicity assay and measurement of the β#x03B2;-actin levels in the cell media were performed as a control to demonstrate the absence of cell membrane rupture with protein release to the cell media ( Fig. 1C–E).
Recently, USP19 has been proposed to regulate protein secretion [29]. To investigate whether MAPS was involved in the release of aSyn, Tau, or Htt, we co-expressed USP19 or the catalytic inactive form USP19 C506 S with aSyn, Tau, or Htt using the HEK stable cell lines we generated above (Supplementary Fig. 2). USP19 significantly increased the secretion of 25QHtt-EGFP; however, the effects did not reach statistical significance for 103QHtt-EGFP, aSyn-EGFP, and EGFP-Tau (Supplementary Fig. 2A). We also tested whether the secreted proteins could be internalized by naüve cells. For this, the cell media from the different stable cell lines was collected and added to naüve HEK cells. Count of the EGFP-positive cells using flow cytometry demonstrated a higher percentage of EGFP-Tau positive cells when compared with the other conditions (Supplementary Fig. 2B). Strikingly, EGFP-Tau was not the protein released at higher levels to the cell media.
These results demonstrate that aSyn, Tau, and Htt are released to the extracellular space and the levels of protein secretion are not directly correlated with the internalization of the proteins by receiving cells and may, instead, depend on their intrinsic properties and on the pathways involved in protein uptake.
Disease-related proteins are present at higher levels in ectosomes
Previous research indicates that aSyn, Tau, and Htt can be secreted in association with EVs [27]. To assess the contribution of ectosomes and exosomes towards the transfer of aSyn, Tau, and Htt, we used an optimized differential ultracentrifugation protocol to purify these EVs from the cell media of HEK cells stably expressing the different proteins (Fig. 2, Supplementary Fig. 3) [28]. The characterization of each vesicle fraction was performed in a previous study [28]. Staining of ectosomal and exosomal fractions showed a similar total protein profile that, as expected, was distinct from that of the whole-cell lysate (Supplementary Fig. 3A). EM imaging confirmed the greater diameter of ectosomes in comparison to exosomes, and their characteristic cup-shape derived from the ultracentrifugation protocol (Supplementary Fig. 3B). In addition, the size distribution and concentration of the two EV types was further assessed using Nanosight (Supplementary Fig. 3C). While ectosomes presented a diameter of 140 nm, the diameter of exosomes was 60 nm. Conventional exosomal protein markers such as alix, flotillin-1, or TSG101 were clearly enriched in the exosomal fraction, whereas the ectosomal fraction was enriched in annexin-A2 and annexin-A5 (evaluated using immunoblot and mass spectrometry) (Fig. 2A, Supplementary Fig. 3D) [28]. The ER and Golgi markers calnexin and GM130, respectively, were not detected, confirming the high purity of the isolated EVs (Supplementary Fig. 3D).
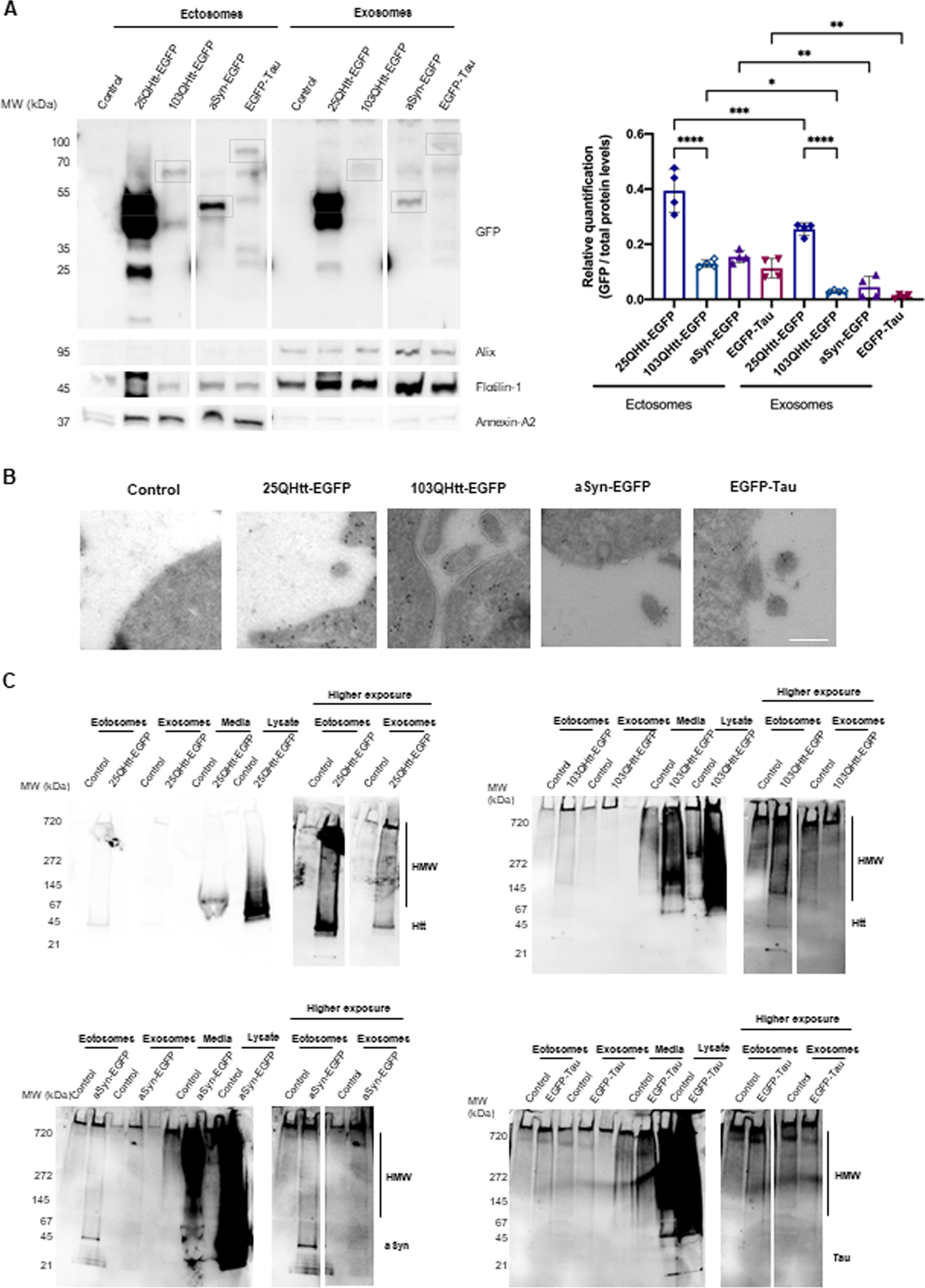
Disease-related proteins are enriched in ectosomes. A) Immunoblots of ectosomal and exosomal fractions purified from the media of HEK cells stably expressing 25QHtt-EGFP, 103QHtt-EGFP, aSyn-EGFP, or EGFP-Tau for 24 h. Equal amounts of protein were separated on SDS-PAGE gel, and membranes were incubated with the indicated antibodies. Protein levels were normalized to total protein levels using Memcode (n = 4). Grey boxes highlight the expected MW (kDa) for each protein. Immunoblots were cropped for space purposes. B) Representative immunoelectron microscopy images of HEK cells stably expressing 25QHtt-EGFP, 103QHtt-EGFP, aSyn-EGFP, or EGFP-Tau immunolabelled for EGFP (scale bar 200 nm). C) Ectosomes and exosomes contain high molecular weight (HMW) species. Ectosomal and exosomal fractions, cell media, and whole-cell lysates of HEK cells stably expressing 25QHtt-EGFP, 103QHtt-EGFP, aSyn-EGFP, or EGFP-Tau. Equal amounts of the proteins were separated on native gels, and membranes were incubated with the indicated antibodies (n = 3). Significant differences were assessed by one-way ANOVA followed by multiple comparisons with significance between groups corrected by Bonferroni procedure. Differences were considered to be significant for values of p < 0.05 and are expressed as mean±SD, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Interestingly, aSyn, Tau, and Htt were detected at higher levels in ectosomes than in exosomes (EGFP levels were normalized to the total protein levels in the immunoblot using MemCode) (Fig. 2A). These results were further confirmed using antibodies specific for aSyn, Tau, or Htt, and by mass spectrometry analyses (Supplementary Fig. 4A-B). We also detected S129 phosphorylation of aSyn, a posttranslational modification (PTM) typically associated with pathology, in the lysates of aSyn-EGFP expressing cells, but not in the EV fractions (Supplementary Fig. 4A). Surprisingly, ectosomes and exosomes containing 25QHtt-EGFP, 103QHtt-EGFP, aSyn-EGFP, or EGFP-Tau presented similar proteomic signatures when compared with EVs purified from control cells (Supplementary Fig. 4C-D).
Immuno-EM experiments demonstrated the presence of the different disease-related proteins in the cytoplasm, as expected, but also near to the plasma membrane, implying their availability to be incorporated in ectosomes (Fig. 2B). Next, to assess the biochemical state of aSyn, Tau, and Htt in the cell media and in EVs, the different samples were applied onto a native gel (Fig. 2C). Overall, cell media presented greater levels of high molecular species when compared with the EV fractions, possibly due to the higher levels of 25QHtt-EGFP, 103QHtt-EGFP, aSyn-EGFP, or EGFP-Tau present in the cell media. Overall, ectosomes containing the disease-related proteins presented a stronger smear when compared with exosomes.
These results highlight the prominent role ectosomes, and not only exosomes, may play in the release of disease-related proteins to the extracellular space.
EVs modify spontaneous activity in primary cortical cultures
Since EVs are actively released to the extracellular space by different types of brain cells, we hypothesized that spontaneous neuronal activity might be influenced by the internalization of ectosomes and/or exosomes in neuronal cells [28].
We treated primary cortical neuronal cultures with 20μ#x03BC;g/mL of ectosomes or exosomes purified from cells expressing 25QHtt-EGFP, 103QHtt-EGFP, aSyn-EGFP or EGFP-Tau, at DIV14, for 24 h to allow internalization of EVs (Fig. 3, Supplementary Fig. 5). We observed internalization of ectosomes and exosomes at similar levels, and that the EV signal was surrounded by LC3 staining, suggesting the targeting of the EVs for degradation after uptake (Fig. 3B, Supplementary Fig. 5A). The internalization of EVs did not cause cell toxicity or induce significant differences in synaptic and autophagic protein levels (Fig. 3C, Supplementary Fig. 5B).
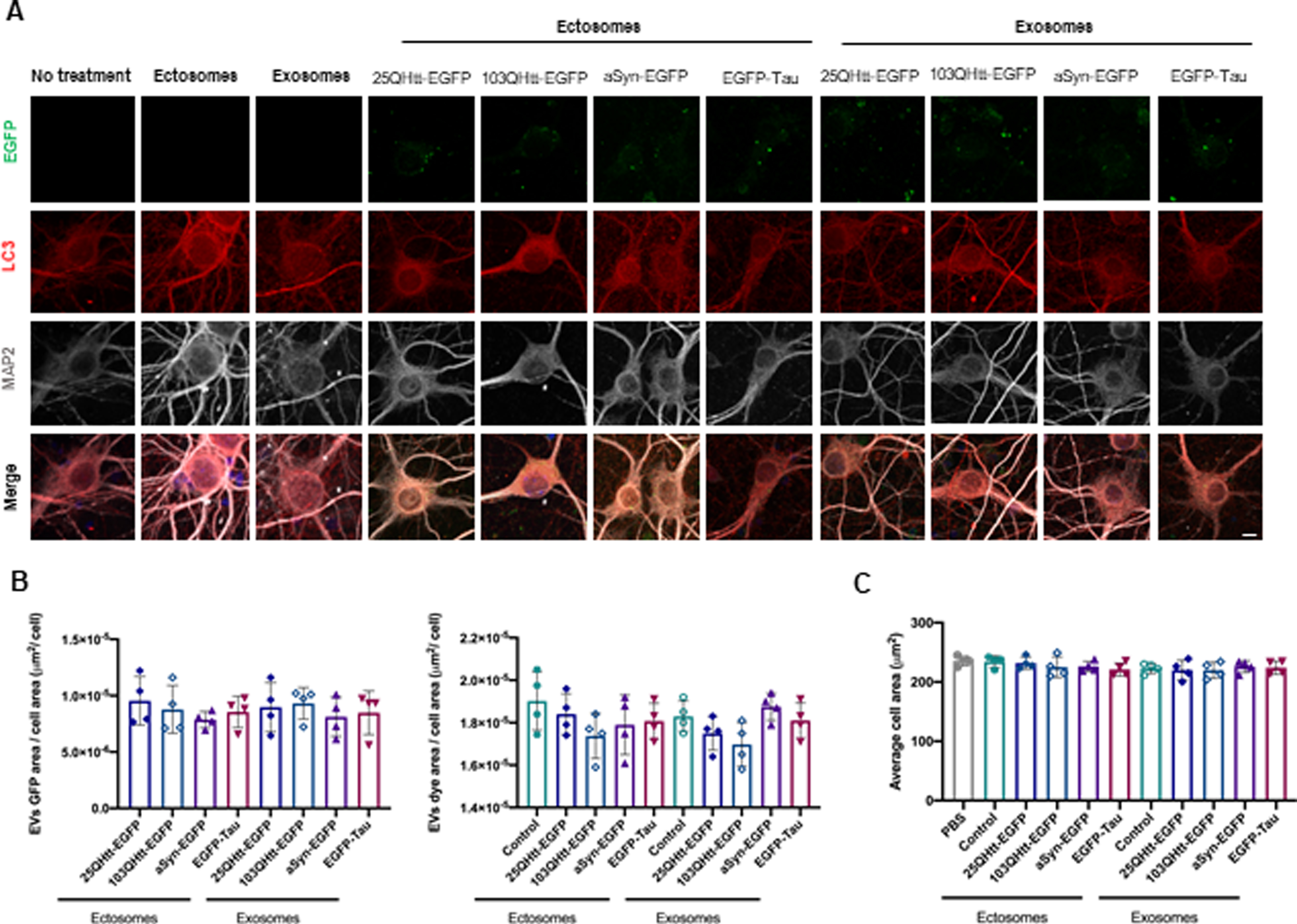
Ectosomes and exosomes containing disease-related proteins are internalized by primary cortical neurons. A) Ectosomes and exosomes from cells expressing 25QHtt-EGFP, 103QHtt-EGFP, aSyn-EGFP, or EGFP-Tau were applied to primary cortical neurons 20μ#x03BC;g/mL for 24 h. Cells were immunoassayed for LC3 (red) and MAP2 (grey). Scale bar 5μ#x03BC;m. B) EV internalization was evaluated through imaging analysis by measuring EGFP signal, dye area, and cell area (n = 4). (C) Neuronal cells treated with EVs do not change average cell area (n = 4). Significant differences were assessed by one-way ANOVA followed by multiple comparisons with significance between groups corrected by Bonferroni procedure. Differences were considered to be significant for values of p < 0.05 and are expressed as mean±SD.
Next, we assessed the effects induced by the EVs on neuronal activity using MEAs [28]. Primary cortical neurons were cultured in MEA chambers and treated with 20μ#x03BC;g/mL of EVs at DIV14. Firing activity was recorded 24 h after treatment with ectosomes or exosomes purified from cells expressing 25QHtt-EGFP, 103QHtt-EGFP, aSyn-EGFP, or EGFP-Tau (Fig. 4). Representative raster plots showed the individual firing activity and bursts events in neuronal cultures for each treatment condition (Fig. 4A). We observed a reduction in the mean firing rate after EV internalization, and a decrease in the average spike amplitude for the neurons treated with exosomes (Fig. 4B). The assessment of bursting activity parameters showed that, upon treatment with EVs, spike bursts were more irregular and with longer duration (Fig. 4C). Overall, exosome internalization resulted in longer inter-burst intervals with reduced intra-burst spiking frequency, and a reduced percentage of spikes within bursts, in contrast to what we observed in neurons treated with ectosomes. Furthermore, these alterations were more strongly correlated with the EV subtype, and not with the presence of aSyn, Tau, or Htt.
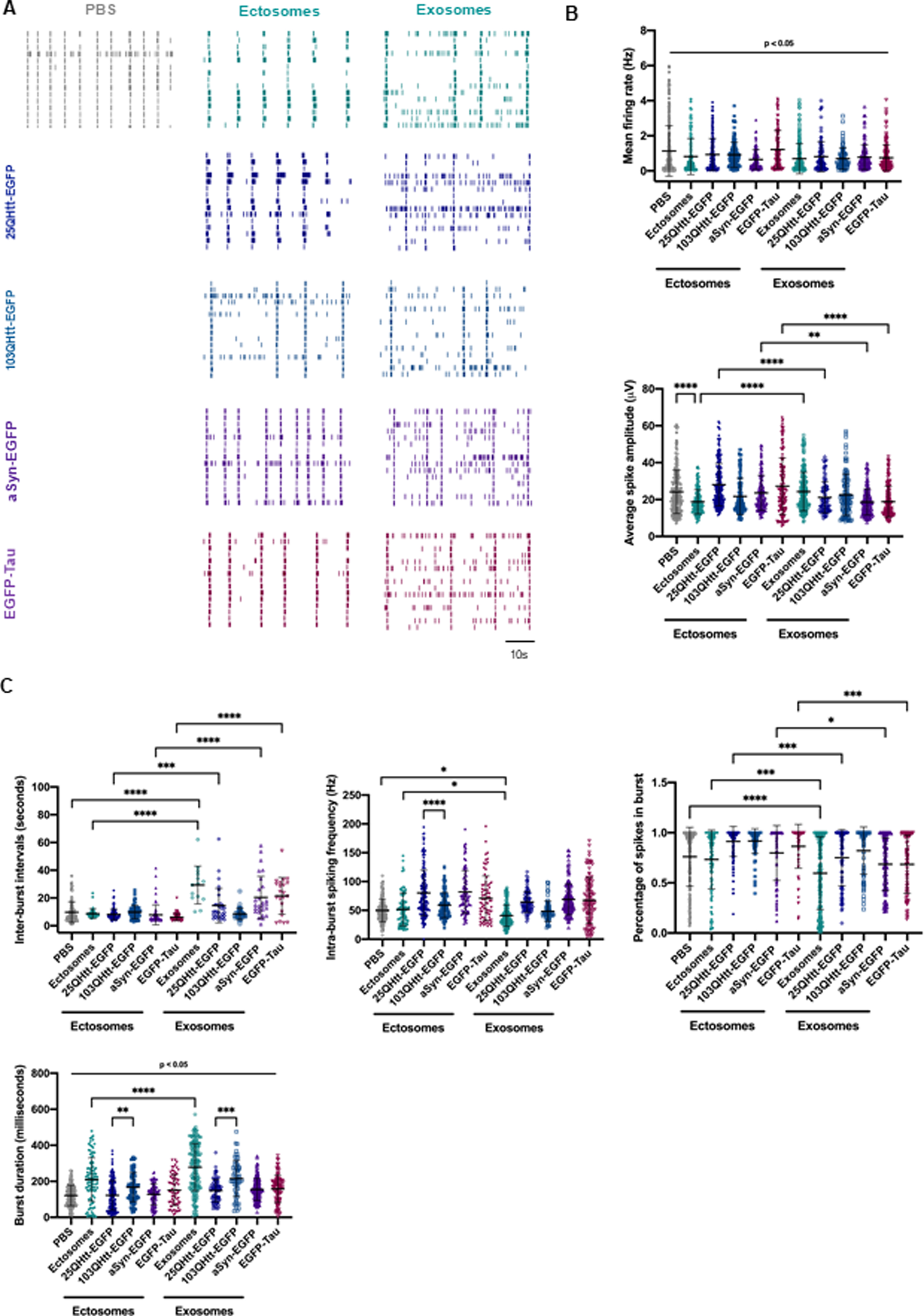
EVs containing disease-related proteins modulate spontaneous activity in primary cortical neurons. A) Representative raster plots of the spontaneous firing activities recorded from cortical neurons after incubation with 20μ#x03BC;g/mL ectosomes and exosomes control or containing aSyn-EGFP, EGFP-Tau, 25QHtt-EGFP, and 103QHtt-EGFP at DIV14 for 24 h, recorded using 60-electrode MEAs. In every block, each row represents one single cell (15 cells shown), and each vertical line represents a single spike obtained on DIV15 [scale bar represents 10 s]. B) Quantification of the mean firing rate and average spike amplitude from primary cortical neurons incubated with PBS or disease-related proteins (n = 3). C) Bursting properties of the cortical neurons treated with PBS or EVs (inter-burst intervals, intra-burst spiking frequency, percentage of spikes in bursts and burst duration) (n = 3). Significant differences were assessed by one-way ANOVA followed by multiple comparisons with significance between groups corrected by Bonferroni procedure. Differences were considered to be significant for values of p < 0.05 and are expressed as mean±SD, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Altogether, our results indicate that, after exposure to EVs, the firing of cultured neurons is more irregular, highlighting the potential of ectosomes and exosomes in modifying important aspects of spontaneous neuronal activity.
EVs are internalized by glial cells and induce the production of pro-inflammatory cytokines
Microglial and astrocytic cells produce neuroinflammatory cytokines and appear to be involved in the spreading in neurodegenerative diseases [51]. Therefore, we then asked whether ectosomes and exosomes containing aSyn, Tau and Htt were internalized by different brain cell types, and whether the disease-associated protein present in the vesicles would alter the responses elicited (Figs. 5 and 6).
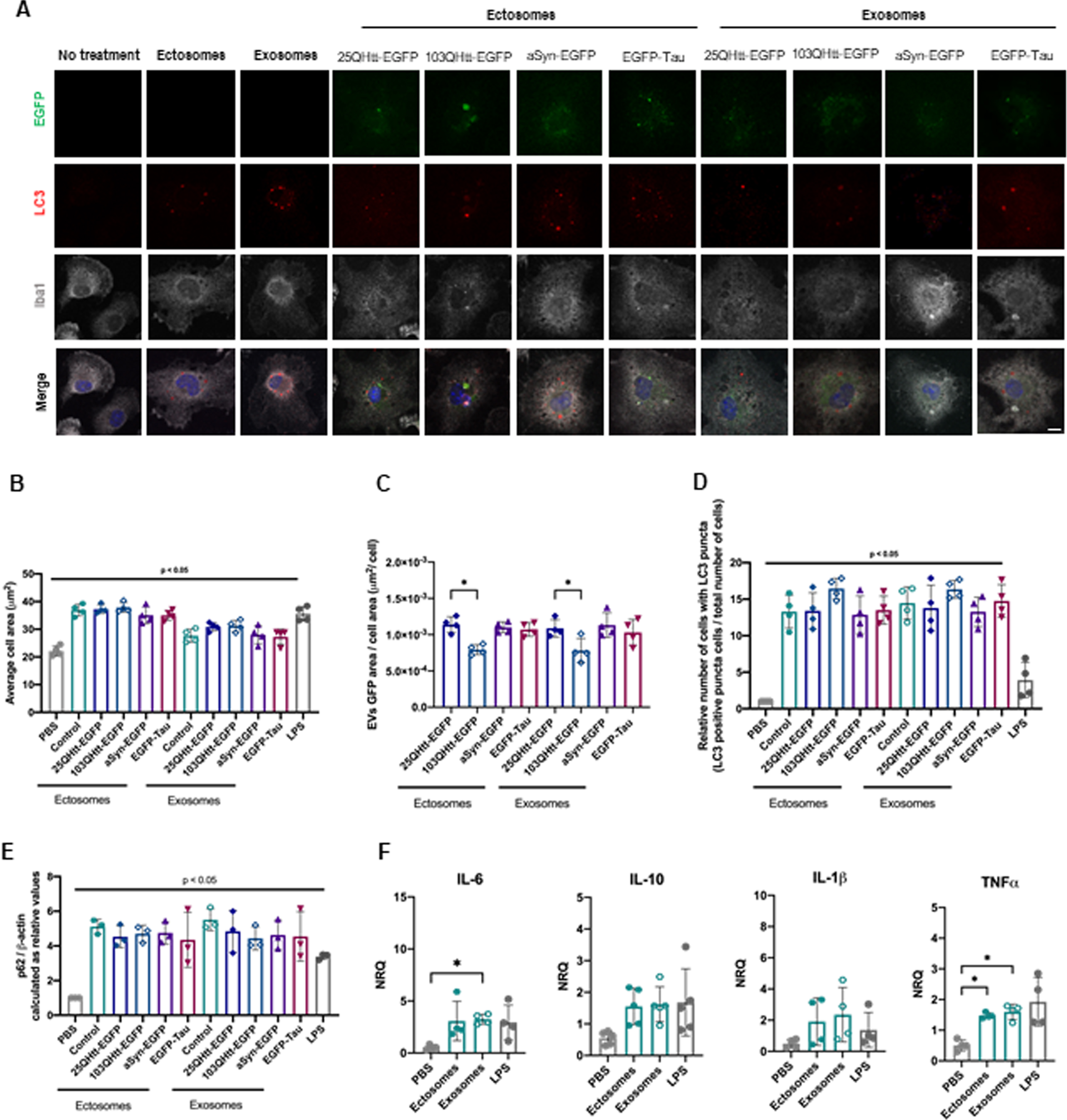
Ectosomes and exosomes containing disease-related proteins are internalized by microglial cells. A) Ectosomes and exosomes from cells expressing 25QHtt-EGFP, 103QHtt-EGFP, aSyn-EGFP, or EGFP-Tau were applied to microglial cultures at a concentration of 10μ#x03BC;g/mL for 24 h. Cells were immunoassayed for LC3 (red) and Iba1 (grey). Scale bar 10μ#x03BC;m. B) Microglial cells treated with EVs show an increase in average cell area (n = 4). C) EV internalization was evaluated through imaging analysis by measuring EGFP signal and cell area. D) Increase in the number of cells containing LC3-positive puncta after EVs treatment (n = 4). E) Increase in p62 protein levels after 24 h of EV treatment. Quantifications were normalized to β#x03B2;-actin levels (n = 3). F) EV treatment resulted in the activation of the pro-inflammatory markers IL-6 and TNFα#x03B1; in microglia cells after 24 h (n = 4). Significant differences were assessed by one-way ANOVA followed by multiple comparisons with significance between groups corrected by Bonferroni procedure. Differences were considered to be significant for values of p < 0.05 and are expressed as mean±SD, *p < 0.05, **p < 0.01.
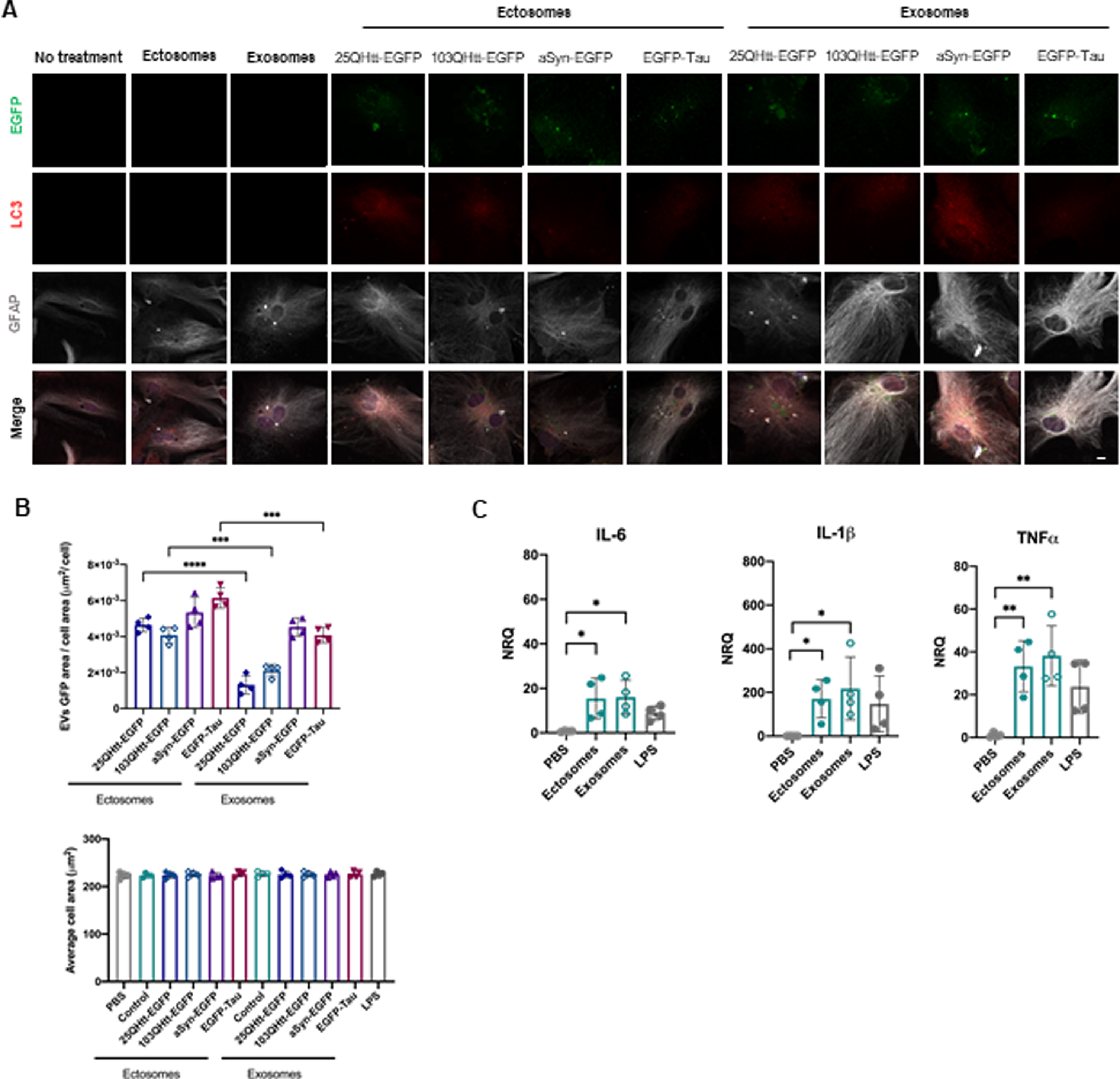
Ectosomes and exosomes containing disease-related proteins are internalized by astrocytic cells. A) Ectosomes and exosomes from cells expressing 25QHtt-EGFP, 103QHtt-EGFP, aSyn-EGFP, or EGFP-Tau were applied to astrocytic cultures at a concentration of 20μ#x03BC;g/mL for 24 h. Cells were immunoassayed for LC3 (red) and GFAP (grey). Scale bar 10μ#x03BC;m. B) EVs internalization levels were evaluated through imaging analysis by measuring EGFP signal and cell area. EVs internalization does not change average cell area (n = 4). C) EV treatment resulted in the activation of the pro-inflammatory markers IL-6, IL-β#x03B2;, and TNFα#x03B1; in astrocytic cells after 24 h (n = 4). Significant differences were assessed by one-way ANOVA followed by multiple comparisons with significance between groups corrected by Bonferroni procedure. Differences were considered to be significant for values of p < 0.05 and are expressed as mean±SD, *p < 0.05, **p < 0.01, ***p < 0.001.
Primary microglial cells were treated with 10μ#x03BC;g/mL of ectosomes and exosomes purified from the media of cells expressing 25QHtt-EGFP, 103QHtt-EGFP, aSyn-EGFP, or EGFP-Tau (Fig. 5, Supplementary Fig. 6). Furthermore, cells were also exposed to the bacterial endotoxin lipopolysaccharide (LPS) as a pro-inflammatory stimulus, as a control [52]. After 24 h of treatment, EGFP-labelled ectosomes and exosomes were found to colocalize with the microglial marker Iba1 in the cell cytoplasm, indicating EVs were internalized (Fig. 5A, Supplementary Fig. 6A). Overall, the internalization ratio was similar for ectosomes and exosomes, independent of the presence of aSyn, Tau, or Htt in the vesicles (Fig. 5C). Interestingly, EVs containing 103QHtt-EGFP were less internalized, or degraded more rapidly, when compared with EVs containing 25QHtt-EGFP (Fig. 5C). EV uptake resulted in microglia activation, as demonstrated by an increase in cell area and elevated levels of the pro-inflammatory cytokines IL-6 and TNFα#x03B1; (Fig. 5B, F). Furthermore, microglial cells displayed an increase in autophagosomes in the cytoplasm (increase in LC3 puncta) and higher p62 levels, indicating autophagy activation (Fig. 5D, E) [53].
To further explore the effect of EV internalization in glial cells, primary astrocytes were treated with 20μ#x03BC;g/mL of ectosomes and exosomes purified from cells expressing 25QHtt-EGFP, 103QHtt-EGFP, aSyn-EGFP, or EGFP-Tau (Fig. 6, Supplementary Fig. 7). After 24 h of treatment, we observed the colocalization of the astrocytic marker GFAP with EGFP-labelled ectosomes and exosomes, confirming internalization (Fig. 6A, Supplementary Fig. 7A). After internalization, the EGFP signal of the EVs was surrounded by LC3 signal, suggesting a possible engulfment and degradation of EVs in the cells. Quantification of the internalization ratio indicated greater engulfment of ectosomes than exosomes, independent of the presence of aSyn, Tau, or Htt in the vesicles (Fig. 6B). Internalization of ectosomes and exosomes caused an increase in pro-inflammatory cytokines, including IL-6, IL-1β#x03B2;, and TNFα#x03B1;, without changing the average cell area (Fig. 6B, C).
These results indicate that different types of EVs can be internalized in microglial and astrocytic cells, independently of the presence of aSyn, Tau and Htt in the vesicles. Furthermore, EV internalization elicited similar responses, with an increase in inflammatory markers and autophagy activation.
Neuronal activity is differentially modulated by the internalization of monomeric aSyn, Tau, or Htt
Our results demonstrate the release of wild-type and pathogenic forms of disease-related proteins to the extracellular space. Although these proteins are taken up by cells, it is still not known whether and how they modulate neuronal activity [54, 55]. To address the functional effects of free forms of aSyn, Tau, and Htt, we treated primary cortical neurons with 100 nM of monomeric aSyn, Tau, 23QHtt, or 43QHtt for 5 days, starting at DIV14 (this concentration was selected as it demonstrated no detrimental effects, which would be incompatible with long-term observations) (Fig. 7) [28].
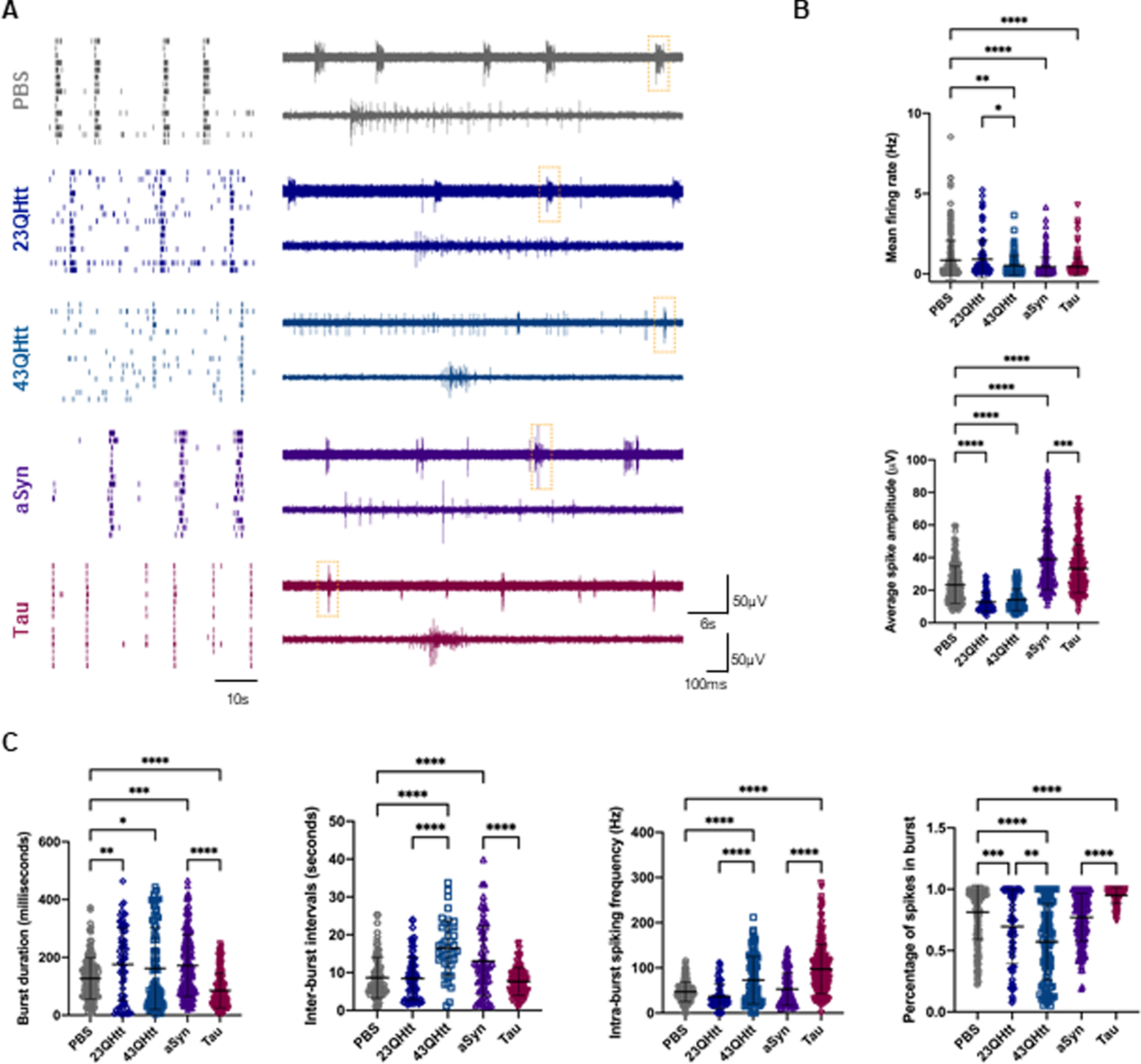
Free monomeric disease-related proteins modulate neuronal activity in primary cortical neurons. A) On the left, representative raster plots of the spontaneous firing activities recorded from cortical neurons after incubation with 100 nM of recombinant protein at DIV14 for 5 days, recorded using 60-electrode MEAs. In every block, each row represents one single cell (15 cells shown) and each vertical line represents a single spike obtained on DIV19 [scale bar represents 10 s]. On the right, representative voltage traces showing the typical firing activity and bursts events in neuronal cultures treated with PBS, monomeric 23QHtt, 43QHtt, aSyn, and Tau (upper traces, scale bars represent 60μ#x03BC;V and 6 s). Closeups of the dashed boxes represent the spikes occurring within a burst ([lower traces, scale bars represent 60μ#x03BC;V and 100 ms]. B) Quantification of the mean firing rate and average spike amplitude from primary cortical neurons incubated with PBS or disease-related proteins (n = 3). C) Bursting properties of the cortical neurons treated with PBS or the disease-related proteins (burst duration, inter-burst intervals, intra-burst spiking frequency and percentage of spikes in bursts) (n = 3). Significant differences were assessed by one-way ANOVA followed by multiple comparisons with significance between groups corrected by Bonferroni procedure. Differences were considered to be significant for values of p < 0.05 and are expressed as mean±SD, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Representative raster plots and voltage traces show the firing activity and burst events in neuronal cultures treated with PBS (as a negative control), 23QHtt, 43QHtt, aSyn, or full-length Tau (Fig. 7A). Overall, neurons treated with monomeric proteins showed a reduction in the mean firing rate. In addition, neurons treated with Htt exhibited a decrease in the average spike amplitude (Fig. 7B). Remarkably, treatment of neuronal cultures with monomeric aSyn or Tau resulted in an increase in the average spike amplitude (Fig. 7B). Furthermore, we observed that Tau induced condensed and more intense spike bursting, as demonstrated by bursts with shorter duration and higher intra-burst spike frequency. Also, the percentage of spikes in each burst increased, suggesting that neurons fire in a more regular and synchronized manner (Fig. 7C). Cells treated with recombinant aSyn exhibited slight alterations in their activity, with longer burst duration and larger intervals between them. Treatment with monomeric 23QHtt or 43QHtt resulted in bursts with longer intervals and a reduction in the number of spikes within the burst when compared with the control. Interestingly, 43QHtt induced a stronger alteration in the spontaneous neuronal activity with a slight reduction in burst rate and a decrease in the percentage of spikes per burst when compared with 23QHtt or PBS-treated cells. In addition, neurons displayed longer inter-burst intervals and higher intra-spike frequency, which was possibly correlated with toxic effects induced by the polyglutamine expansion.
Altogether, our results demonstrate that different disease-related proteins induce specific effects on spontaneous neuronal activity.
DISCUSSION
Several neurodegenerative disease-related proteins appear to be transferred from cell-to-cell, contributing for the spreading of pathology and disease progression [19–21]. However, the basic molecular mechanisms involved in the release of proteins which are, oftentimes, not typical secretory proteins, are still unclear. Likewise, the effect of such proteins once they are in the extracellular milieu, is also unclear. Therefore, deciphering the pathways through which brain cells release, sense, and respond to the extracellular presence of normal and pathological forms of disease-associated proteins is essential. Here, we conducted a systematic comparison of the basic molecular mechanisms involved in the release of three proteins associated with distinct neurodegenerative disorders, aSyn, Tau, and Htt, and of general cell-autonomous responses in different brain cell types. Since these proteins accumulate in different types of brain cells, forming distinct proteinaceous inclusions, and spreading through different neuronal circuits, it is important to establish differences and similarities in the ways they are handled to identify specific therapeutic targets for each disease. In our study, exploiting simple, yet tractable, cell systems, we found that aSyn, Tau, and Htt are transferred between cells at different levels, but using overlapping cellular pathways. Importantly, we report that the release of these proteins in a free form, or in EVs, elicits different molecular processes in neighboring cells.
By taking advantage of stable cell lines expressing aSyn, Tau, 25QHtt, and 103QHtt fused to EGFP, as a common denominator, we were able to study and compare different molecular mechanisms involved in the release, and uptake of the various in receptor cells. We demonstrate that aSyn, Tau, and Htt are released to the extracellular space at different levels. Importantly, the presence of the EGFP tag did not alter the release patterns of these proteins.
Our study further indicates that the MAPS pathway might modulate the release of a fraction of aSyn, Tau, or Htt. In agreement with previous studies [29], USP19 expression in our stable cell lines led to a slight increase in the release of aSyn, Tau, and Htt. Interestingly, USP19 promoted a significant increase in the secretion of 25QHtt-EGFP, suggesting a relevant role for this pathway in HD, as it was previously described to interact and regulate mutant Htt protein levels and promote its aggregation [56, 57].
After release, aSyn, Tau, or Htt were found to be internalized by naüve receptor cells. In particular, we observed a significant increase in the percentage of EGFP-positive cells after incubation of naüve cells with media collected from cells expressing EGFP-Tau. Although 25QHtt-EGFP and aSyn-EGFP were more abundant in the cell media than EGFP-Tau, we observed more internalization of this protein in cells. These results suggest that Tau might be more easily, or more rapidly, internalized by cells, or that different pathways might be involved in its uptake when compared with the other proteins. Strikingly, this observation also demonstrates that protein internalization is not a process solely dependent of the protein levels in the exterior space but determined by the type of protein and mechanisms involved in the cellular uptake.
In addition to the release of proteins in free form, several studies have also reported the secretion of disease-related proteins in ectosomes and exosomes [27, 58]. Exosomes are the most extensively studied type of EVs, and are also implicated in the secretion of pathological proteins [59]. However, Tau was also found to be present in ectosomes extracted from culture media from cell models and human cerebrospinal fluid [58, 60], highlighting the relevance of this EV type, and need for further research to address their role in neurodegenerative diseases. In our study, we found that ectosomes purified from the cell media of stable cell lines contained higher levels of aSyn, Tau, and Htt than exosomes. Interestingly, we observed that aSyn, Tau, and Htt are present near the plasma membrane, in agreement with their possible incorporation in ectosomes, and release via passive diffusion. Traditionally, ectosome characterization has been challenging due to the lack of specific protein markers. In our study, we highlight the enrichment of annexin-A2 in ectosomes, suggesting this protein as a specific marker for this EV type, as previously described [28].
We also found that incorporation of aSyn, Tau, or Htt in EVs did not change the normal vesicle protein composition in mass spectrometry analyses, suggesting these vesicles are not deregulated when they transport the disease-associated proteins. However, future research will be necessary to determine whether the content of other biomolecules, such as lipids or nucleic acids, is altered, and this then inform on whether the uptake of the EVs by receptor cells may be altered. This is particularly relevant in the context of neurodegenerative diseases, as these vesicles may not only play a role in the transmission of proteins but also in signaling cellular alterations taking place during the disease process. In PD, in particular, investigating the internalization of different types of vesicles, as we have done here, will impact on the way we envision disease-modifying therapies, and also in the field of disease biomarkers, since different EV types may turn out to hold different value for monitoring disease progression.
Microglia and astrocytes are two important cell types in the brain that mediate neuroinflammatory processes and, thereby, playing an important role in the pathogenesis of neurodegenerative diseases [61]. Our study shows that the uptake ectosomes and exosomes can be taken up by microglial and astrocytic cells, and that both EV types elicited an increase in pro-inflammatory cytokines (IL-6, IL-1β#x03B2;, and TNFα#x03B1;). Microglial cells adopted an activated phenotype and exhibited LC3 puncta in the cytoplasm and increase in p62 levels, indicating autophagy activation, possible for the clearance of the EVs. Astrocytes also displayed accumulation of EVs in the cytoplasm, with the EGFP signal being surrounded by LC3 staining. Interestingly, microglia cells were more sensitive to EV-treatment: while astrocytes tolerated 20μ#x03BC;g/mL, microglia tolerated only up to 10μ#x03BC;g/mL of EV protein. Ectosomes and exosomes containing aSyn-EGFP, EGFP-Tau, or 25QHtt-EGFP were taken up at similar levels in microglia, and these were higher than those observed with 103QHtt-EGFP. Indeed, both wild-type and mutant Htt can influence vesicle transport in the secretory and endocytic pathways through associations with clathrin-coated vesicles [62]. In contrast, astrocytes seemed to, in general, internalize more ectosomes, or degrade exosomes faster. Together, our findings demonstrate that EVs can be targeted by different types of glial cells, and that their uptake and effects are likely correlated with the EV type and content. These findings provide new insights into molecular mechanisms of intercellular communication.
We also observed that ectosomes and exosomes are taken up by primary cortical neurons, as previously described [28]. The internalization ratio was similar for EVs with aSyn, Tau, or Htt, but was considerably lower than that observed with microglia and astrocytes. The potential effects of EVs on neuronal network activity are still unclear. In this context, we demonstrate that spontaneous neuronal function can be modulated by ectosomes and exosomes, and that EV internalization is associated with a disruption of the typical synchronized bursting activity, resulting mostly in lower and less organized spiking activity. Interestingly, these alterations are mainly correlated with the EV subtype, and not with the presence of aSyn, Tau, or Htt, although slight differences could be perceived between 25QHtt-EGFP and 103QHtt-EGFP [28].
We also demonstrate that aSyn, Tau, or Htt present in the cell media can modify the spontaneous activity in cortical neurons. The use of identical concentrations of monomeric protein allowed us to compare the consequences of aSyn, Tau, and Htt internalization, at sub-cytotoxic concentrations. aSyn function has been associated with synaptic activity through the regulation of the vesicle pool [63, 64]. Interestingly, incubation of cortical neurons with monomeric aSyn resulted in a reduction of the firing rate and in an increase in burst duration, without changing the coordinated network activity. These effects contrast with those reported with higher concentrations of extracellular aSyn or with aggregated assemblies that strongly reduce neuronal activity by disrupting synaptic transmission, thereby contributing to neuronal death [65]. Remarkably, treatment of neurons with monomeric Tau results in increased neuronal activity and in robust and synchronized bursting activity, suggesting that neurons fire in a more regular and synchronized manner. Consistently, full-length monomeric Tau was previously described to be rapidly and efficiently internalized in healthy neurons, implying this might be part of a physiological, and not pathological, process [55]. Treatment of neurons with 23QHtt or 43QHtt resulted in longer burst intervals and a reduction in the synchronized bursting activity. As expected, internalization of 43QHtt resulted in greater impairment in the coordinated network activity, correlated with the toxic effects of the polyglutamine expansion [66–68]. A detailed understanding of the mechanisms of internalization of monomeric and aggregated forms of aSyn, Tau, and Htt will be invaluable for the development of potential therapies for preventing the interneuronal transfer of proteins, without interfering with the physiological transfer of non-pathogenic forms.
Overall, our systematic study compares the transfer of disease-related proteins through various cellular mechanisms, and between different cell types. In particular, we emphasize that protein release, either in a free form or in EVs, induces diverse effects in neighboring receptor cells, and that great care is important when considering the development of therapeutic strategies to avoid interfering with normal physiological intercellular communication.
Footnotes
ACKNOWLEDGMENTS
We thank Prof. Dr. Ray Truant (McMaster University, Ontario, Canada) for kindly providing the Htt antibody used in the immunoblot experiments [anti-N17 (1-8)]. We also thank Prof. Dr. Hilal Lashuel (EPFL, Switzerland), for kindly providing the recombinant 23QHtt and 43QHtt, and to Prof. Dr. Flaviano Giorgini (Leicester University, Leicester, United Kingdom) for providing the Htt lentiviral vectors. We thank Sabine König and Uwe Plessmann from Max Planck Institute for Biophysical Chemistry (Göttingen, Germany) and Christof Lenz from the Core Facility Proteomics at University Medical Center Göttingen (Göttingen, Germany) for assisting with mass spectrometry analysis. We thank Dr. Nicolés Lemus (Göttingen, Germany) for support with flow cytometry experiments.
T.F.O was supported by European Union’s Horizon 2020 research and innovation program under grant agreement No. 721802 (SynDegen), and by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy –EXC 2067/1-390729940, by SFB1286 (B8). T.G. is supported by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement number 724822). This work was partly supported by the Göttingen Graduate School for Neurosciences, Biophysics, and Molecular Biosciences (DFG grant GSC 226/4).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
All data are available upon request. For further inquiries, please contact the corresponding author Prof. Dr. Tiago Fleming Outeiro (touteir@gwdg.de).