
Abstract
DJ-1 mutations are rare causes of autosomal recessive early-onset Parkinson’s disease (AR-EOPD) and relatively rarely reported in the Chinese population. Here, we used the whole-exome sequencing and Sanger sequencing to investigate DJ-1 mutations in the Chinese population and confirmed the pathogenicity of the mutation using primary fibroblasts established from skin biopsies. We identified a novel homozygous mutation (c.390delA, p.D131Tfs*3) in DJ-1 in a consanguineous Chinese family. The proband in this family had parkinsonism at the age of 22. His brain MRI indicated brain iron accumulation in the basal ganglia and cerebellum. The novel mutation caused DJ-1 protein deficiency, led to mitochondrial dysfunction, inhibited cell proliferation, and anti-oxidant defense.
INTRODUCTION
Parkinson’s disease (PD) is a common neurodegenerative disease clinically characterized by classic motor symptoms and other non-motor symptoms [1]. Most PD patients are sporadic, only 5-10% of patients carry causative mutations [2]. The DJ-1 mutation is one of the causative mutations associated with autosomal recessive early-onset PD (AR-EOPD), as an occurrence of PD before the age of 40 years [3]. More than 40 DJ-1 mutations have been reported, but the DJ-1 mutations were relatively rare in Chinese patients with AR-EOPD [3–5]. Although most of these patients had typical phenotypes, they also presented some heterogeneity, such as dementia [6, 7], amyotrophic lateral sclerosis-like symptoms [6, 9], and distal spinal amyotrophy [10] (Supplementary Table 1). Here we reported a consanguineous Chinese family with early-onset parkinsonism and brain iron accumulation carrying a novel DJ-1 mutation (c.390delA, p.D131Tfs*3), and investigated the pathogenicity of this mutation in cultured human skin fibroblasts.
METHODS
Subjects
The study was approved by Second Affiliated Hospital, Zhejiang University School of Medicine. Blood was collected from all family members after obtaining informed consents. The family consists of 3 off-springs, of whom two were affected. The proband underwent a detailed neurological examination. Routine blood tests and radiological examinations were also performed.
Genetic analysis
DNA was extracted from the peripheral blood using Blood Genomic Extraction Kit (Qiagen, Hilden, Germany). Whole-exome sequencing (WES) was performed in the proband using the Agilent SureSelect Human All Exome V6 kit (Agilent Technologies Inc, Canada) on an Illumina HiSeq X Analyzer (Illumina, USA). Sequence reads were aligned against the human reference genome (UCSC hg19). The ANNOVAR software was performed to annotate the variants. Variants were filtered based on a minor allele frequency < 1% in dbSNP (https://www.ncbi.nlm.nih.gov/snp/), 1000 Genomes Project (https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/), ExAc (https://exac.broad.institute.org/) and gnomAD (https://gnomad.broadinstitute.org/) databases and functional impact on protein change referred to American College of Medical Genetics and Genomics (ACMG) standards and guidelines [11]. Sanger sequencing using primer DJ-1 E6F (5′TTCTACCTAGGCCTCCCC3′) and DJ-1 E6R (5′AGAGAAGAATCGCTT GAACCC3′) confirmed segregation.
Primary fibroblasts culture and treatment
Primary fibroblast cell lines were established from skin biopsies with punches (Electron microscopy sciences, USA), acquired from the proband and normal controls. Written consents were obtained from these subjects. Fibroblasts were maintained in DMEM (GIBCO, USA) with glucose (4.5 g/L) and sodium pyruvate supplemented with 10% FBS (GIBCO, USA) at 37°C in 5% CO2.
RNA isolation and expression detection
The total RNA was extracted from the fibroblasts using Tissue Total RNA Isolation Kit (Vazyme, China). The total RNA was reversely transcripted to cDNA using PrimeScript RT Master Mix (Takara, Japan). Quantitative real-time PCR was performed using TB Green Premix Ex Taq (Tli RNaseH Plus) (Takara, Japan) by means of relative quantification (2–ΔΔCt method). Primer sequences used for quantitative PCR were as follow: DJ-1 F (5′GCTCTGGTCATCCTGGCTAAA3′), DJ-1 R (5′GACAAATGACCACATCACGGC3′); GAPDH F (5′AGATCCCTCCAAAATCAAGTGG3′), GAPDH R (5′GGCAGAGATGATGACCCTTTT 3′).
Western blotting
The cells were harvested in the RIPA Lysis Buffer (Beyotime, China) supplemented with a 1% protease inhibitor PMSF (Beyotime, China) and centrifuged at 13,000 rpm for 20 min at 4°C. Protein extracts were separated on 12% SDS-PAGE gels and transferred to a polyvinylidene fluoride membrane. The following primary antibodies were used: anti-DJ-1 (1:1000, Abclonal A19097, China) and anti-β-actin-HRP (1:10000, HuaBio, China). HRP-conjugated secondary antibody (1:5000, Abclonal, China) was used to detect primary anti-DJ-1 antibody.
Immunofluorescence analysis
Cells were fixed with 4% paraformaldehyde for 5 min at room temperature and blocked in PBS containing 0.3% TritonX-100 and 10% bovine serum albumin for 60 minutes. Cells were then incubated with anti-DJ-1 (1:100, Abclonal A0987, China) antibody in the blocking buffer at 4°C overnight, followed by secondary anti-rabbit IgG Alexa Fluor 596 antibody (1:1000, Life Technologies, USA). Cell nuclei were then stained with 40, 6-diamidino-phenylindole (DAPI; 1:10000, Life Technologies, USA). Fluorescence images were captured by Olympus FV3000 confocal system.
Cell Counting Kit-8 (CCK-8) assay
CCK-8 (Beyotime, China) was used to measure cell proliferation and toxicity. A total of 104 cells in a volume of 100μL per well were cultured in four replicate wells in a 96-well plate in the medium containing 10% FBS. Then, the CCK-8 reagent (10μL) was added to 100μL DMEM to generate a working solution, of which 110μL was added per well and incubated for 2 h. The absorbance was measured at 450 nm.
ATP measurement
ATP levels were measured by an ATP Activity Assay Kit (Solarbio, China) following manufacturer’s instructions. Briefly, a total of 500 million fibroblasts were collected and supernatant was measured at 340 nm. ATP content fold change was the ratio of ATP content (μmol/106 cell) of the proband and the control.
Statistical analysis
Data were presented as the mean±95% confidence interval. The following conventions were used: *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant. Student’s t-test in 2 groups for statistical significance was performed using GraphPad Prism 7 software.
RESULTS
Clinical features
The proband (IV-3) from the consanguineous family (Fig. 1A) was a 27-year-old male who had five years of tremor. He denied any exposure to toxins and radioactive substances. He admitted to smoking four to five cigarettes a day for ten years. At the age of 22 years, he developed a left-hand tremor. Then he began to tremble in bilateral hands, and the symptom of tremor worsened with emotion and upper limb movement. In the beginning, there were no signs of rigidity, bradykinesia, or psychiatric symptoms. He was first seen in our department at the age of 24 years. Initially, he was diagnosed with “somatic symptom disorder” and treated with haloperidol (3 mg/day). But the treatment was ineffective, and the symptoms progressed quickly. A half month after being newly diagnosed, the tremor of both hands was worse, accompanied by bradykinesia and reduced arm swing. He also showed mild anxiety, constipation, and insomnia.
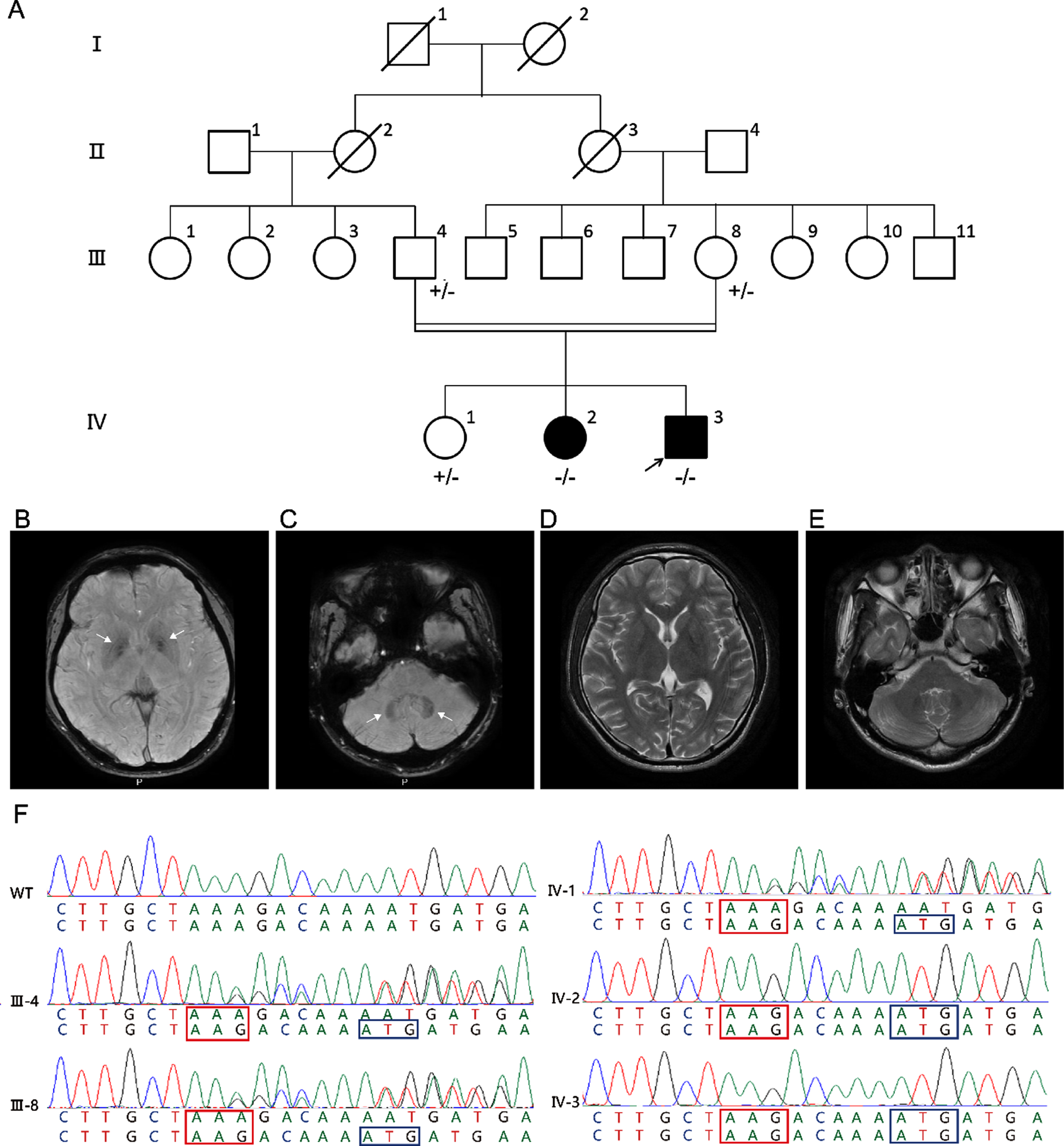
A) Family pedigree. Arrow: proband; black symbols: affected subjects; white symbols: unaffected subjects; –/–: homozygous DJ-1 p.D131Tfs*3 mutation carrier; +/-: heterozygous DJ-1 p.D131Tfs*3 mutation carrier. B-E) Brain MRI of the proband. Susceptibility weighted imaging (SWI) indicates brain iron accumulation in the bilateral globus pallidus (white arrows) (B) and cerebellar dentate nucleus (white arrows) (C). No abnormalities are found on T2- weighted in the bilateral globus pallidus (D) and cerebellar dentate nucleus (E). F) Chromatograms of p.D131Tfs*3 within DJ-1 in proband (IV-3) and family members (III-4, III-8, IV-1 and IV-2). Red box: frameshift triplet codon; blue box: premature stop codon.
Neurological examinations demonstrated the increased muscle tone in the extremities, postural tremor, involuntary movement, and abnormal alternating movements in bilateral hands. Muscle strength, posture, and gait were intact. The score on the part III Unified Parkinson’s Disease Rating Scale (UPDRS) score was 37/104. The tendon reflexes in the lower limbs were brisk. Cognition was intact (Mini-mental State Examination 28/30). Brain magnetic resonance imaging (MRI) indicated brain iron accumulation in the bilateral globus pallidus (Fig. 1B) and cerebellar dentate nucleus (Fig. 1C). No abnormalities were found on T2-weighted in these regions (Fig. 1D, E). The initial treatment was discontinued and replaced with levodopa and benserazide hydrochloride (375 mg/day), amantadine (200 mg/day), and clonazepam (0.5 mg/day). The motor symptoms were ameliorated.
Individual IV-2 (Fig. 1A) was a 29-year-old female. Her mother and brother all recognized that she had hand tremors, and it became severe with emotion. Her symptoms were similar to those of the proband. Unfortunately, we could not get more information about her.
Genetic analysis
After whole-exome sequencing (WES) analysis in the proband, a novel homozygous frameshift variant in DJ-1 (NM_007262.4: c.390delA, p.D131Tfs*3) was identified. This variant was homozygous in both affected offsprings and presented in the heterozygous state in unaffected parents and offspring using Sanger sequencing (Fig. 1F). It caused a frameshift and premature stop codon in exon 6 of DJ-1 and was absent in dbSNP, 1000 Genomes Projects, ExAC, gnomAD, and our WES database with 500 Chinese controls.
Pathogenicity of the mutation
As shown in Fig. 2A, the monomer of DJ-1 protein contains 8 α helices and 11 β folds and tends to form a dimer through interactions of the β3-β4 hairpin, β11-αG loop, and αH helices [12]. The p.D131Tfs*3 mutation caused the destruction of the C-terminus, which was important to stabilize the dimerization of DJ-1. To investigate the basic properties of DJ-1 gene expression in living cells, we cultured skin fibroblasts from the proband and normal controls followed by quantitative real-time PCR. We found that the DJ-1 mRNA level in the proband was significantly decreased compared to the control (Fig. 2B). Western blotting and immunofluorescence with an anti-DJ-1 antibody were used to further confirm the expression of DJ-1 protein. As a result, p.D131Tfs*3 DJ-1 protein was barely detectable (Fig. 2C, D).
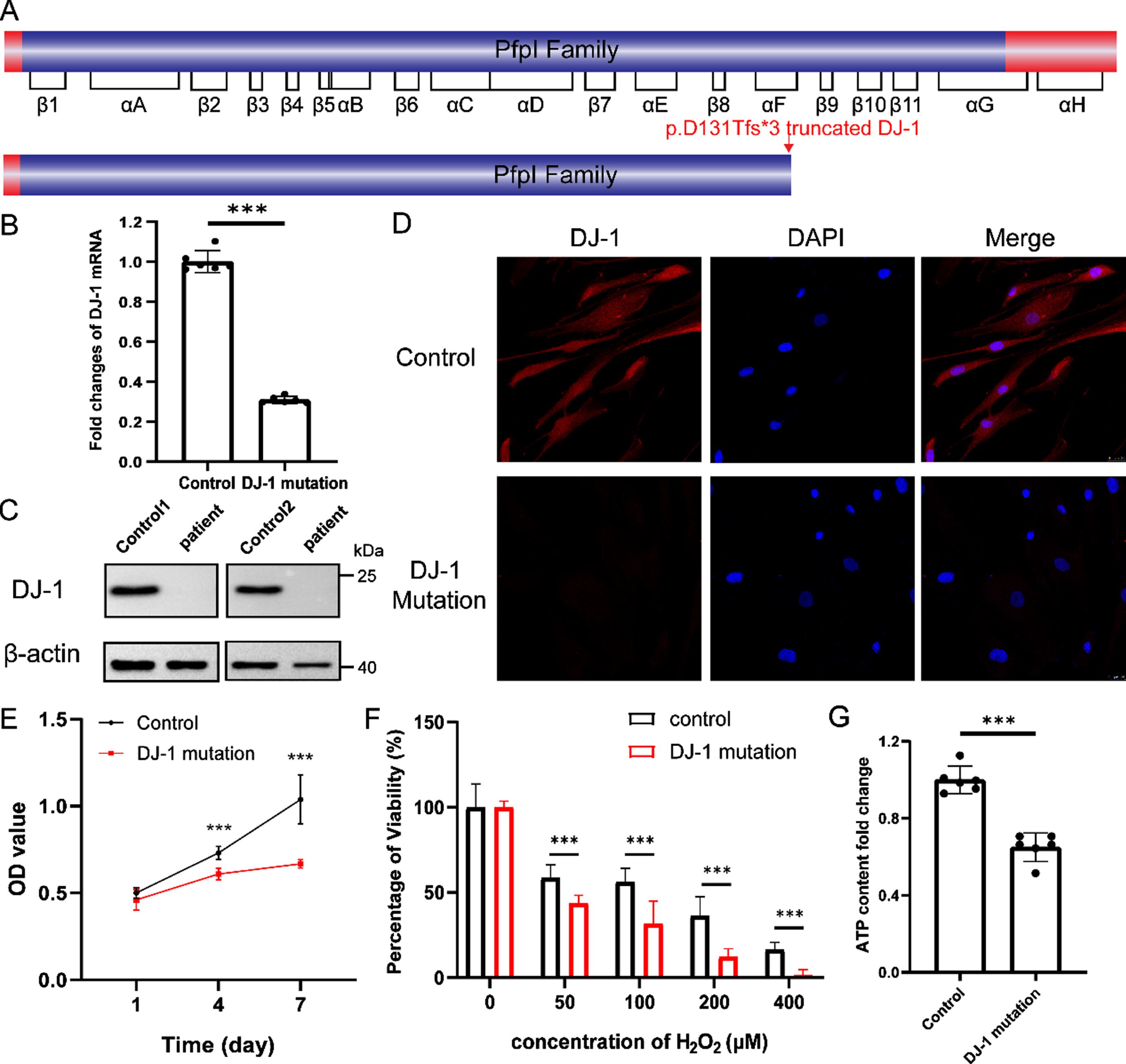
A) A schematic representation of the full-length DJ-1 protein and effect on the protein structure of the variant in the pedigree. B) The fold changes of DJ-1 mRNA expressed in fibroblasts represent mean±95% confidence interval (n = 6, ***p < 0.001). C) Western blot analysis of DJ-1 protein. D) Staining of DJ-1 (red) and DAPI (blue) in fibroblasts from the control and the proband. Scale bar, 25μm. E) Growth curve of fibroblasts. OD values are measured by CCK-8 assay on day 1, 4, and 7, respectively. Values represent mean±95% confidence interval (n = 4, ***p < 0.001). F) Viability of fibroblasts treated with different concentration (0, 50, 100, 200, 400μM) of hydrogen peroxide (H2O2) for 2 h.
Then, we explored the effect of the mutation of DJ-1 on fibroblasts. Firstly, we detected the proliferation of fibroblasts on the day 1, 4, and 7 using Cell Counting Kit-8 (CCK-8) assay. On day 1, there was no significant difference in proliferation between the fibroblasts from the proband and normal control. And the proliferation rate of control fibroblasts was significantly higher than that of the proband on day 4 and day 7 (Fig. 2E). Since DJ-1 protein was reported to be associated with oxidative stress and mitochondrial function [13], we compared the anti-oxidative stress ability of cells between the proband and control. After the treatment with different concentrations of hydrogen peroxide for 2 h, the fibroblasts from the proband showed significantly decreased viability (Fig. 2F). We also detected the ATP content, which reflects mitochondrial function, and found that ATP levels in the proband was significantly reduced (Fig. 2G). Overall, the novel mutation in DJ-1 inhibited cell proliferation, decreased the ability to resist oxidative stress and caused mitochondrial dysfunction.
DISCUSSION
Here we reported a novel homozygous mutation (c.390delA) in exon 6 of DJ-1 segregating with PD in a consanguineous family from China. Among the three major AR-EOPD related genes (PINK1, Parkin, and DJ-1), DJ-1 is the lowest frequency reported, accounting for approximately 1-2% of all EOPD cases [14, 15]. The proband has a common clinical phenotype of DJ-1 mediated PD, such as early onset, unilateral onset, parkinsonism, and several non-motor symptoms [16] (Supplementary Table 1). Moreover, to our knowledge, this is the first time that DJ-1 mutation is identified with brain iron accumulation explicitly. However, the association between iron homeostasis and neurodegeneration is still insufficiently understood [17, 18]. It has been reported that DJ-1 participated in mitochondrial respiratory chain organization [19–21]. Therefore, the novel mutation in our study, which may abolish DJ-1 protein expression, will result in mitochondrial dysfunction. A mass of intracellular iron is utilized for the synthesis of iron-sulfur clusters in mitochondrial complex I [22]. Thus, mitochondrial dysfunction and disorganization caused by DJ-1 mutant may trigger iron dyshomeostasis. Furthermore, Chin et al observed altered iron responsive element (IRE) gene transcript abundance in dj1 knock out zebrafish brain [23]. IRE gene sets are important to maintain iron homeostasis. In turn, iron overload in brain regions can promote oxidative stress [18]. We confirmed that cells with DJ-1 mutation were more sensitive to oxidative damage. This may further cause apoptosis and ferroptosis [24, 25]. Dying neurons in PD that release neuromelanin–iron complexes make the situation worse [26, 27].
Collectively, we reported the first AR-EOPD patient with brain iron accumulation associated with a novel DJ-1 mutation from a consanguineous Chinese family. The DJ-1 mutation may cause a vicious circle between iron dyshomeostasis and oxidative stress. In addition to typical EOPD symptoms, DJ-1 mediated PD presented some heterogeneity in clinical characteristics with different mutants. These atypical features provide new insights into the function of DJ-1 protein and the pathogenesis of neurodegenerative diseases.
Footnotes
ACKNOWLEDGMENTS
The authors are grateful to all subjects for participation in our study.
This study was supported by the grant from the Key Research and Development project of Zhejiang Province (2019C03039) and the research foundation for distinguished scholar of Zhejiang University to Zhi-Ying Wu (188020–193810101/089, Hangzhou).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.