
Abstract
Background:
Braak and others have proposed that Lewy-type α-synucleinopathy in Parkinson’s disease (PD) may arise from an exogenous pathogen that passes across the gastric mucosa and then is retrogradely transported up the vagus nerve to the medulla.
Objective:
We tested this hypothesis by immunohistochemically staining, with a method specific for p-serine 129 α-synuclein (pSyn), stomach and vagus nerve tissue from an autopsy series of 111 normal elderly subjects, 33 with incidental Lewy body disease (ILBD) and 53 with PD.
Methods:
Vagus nerve samples were taken adjacent to the carotid artery in the neck. Stomach samples were taken from the gastric body, midway along the greater curvature. Formalin-fixed paraffin-embedded sections were immunohistochemically stained for pSyn, shown to be highly specific and sensitive for α-synuclein pathology.
Results:
Median disease duration for the PD group was 13 years. In the vagus nerve none of the 111 normal subjects had pSyn in the vagus, while 12/26 ILBD (46%) and 32/36 PD (89%) subjects were pSyn-positive. In the stomach none of the 102 normal subjects had pSyn while 5/30 (17%) ILBD and 42/52 (81%) of PD subjects were pSyn-positive.
Conclusion:
As there was no pSyn in the vagus nerve or stomach of subjects without brain pSyn, these results support initiation of pSyn in the brain. The presence of pSyn in the vagus nerve and stomach of a subset of ILBD cases indicates that synucleinopathy within the peripheral nervous system may occur, within a subset of individuals, at preclinical stages of Lewy body disease.
INTRODUCTION
Since the discovery of α-synuclein as the major constituent of Lewy bodies, sensitive immunohistochemical (IHC) methods have demonstrated that the brain and peripheral nervous system (PNS) distribution and density of Lewy bodies and their associated abnormal neurites are much greater than formerly appreciated [1–4]. Furthermore, the common PNS occurrence of α-synuclein pathology has instigated a proliferation of research aimed at using PNS α-synuclein pathology as a biopsy biomarker as well as raising the critical question as to whether or not it begins in the brain or within the periphery [5–47]. The stimulus for the latter alternative, which might be called the “body-first” hypothesis, has come largely from clinical studies of PD that have found a wide range of non-motor signs and symptoms that accompany or may even precede the motor signs [48–50]. Many of these non-motor accompaniments are related to gastrointestinal (GI) dysfunction and therefore much attention has been focused on the stomach as the major “first stop” along the alimentary tract [43, 52] and hence the most likely place for the initiation of synucleinopathy, perhaps by absorption of toxins or through microbial or inflammatory stimuli, followed by transmission to the brain through the vagus nerve.
Supporting this is the repeatedly-confirmed finding of a rostrocaudal GI gradient of synucleinopathy [1, 31], which may itself be related to the known distribution of vagal GI innervation [53]. A stomach-vagal-brain connection has been invoked to explain reports that subjects with prior vagotomy may have a lower prevalence of PD, although this has been disputed [54–59]. A stomach “inoculation” site has now been experimentally tested in multiple animal models, and a consensus seems to have emerged that in fact bidirectional spread is possible, both upwards from the stomach and downwards from the brainstem, including a possible hematogenous route [60–62].
A major piece of evidence for the body-first hypothesis, however, has been lacking, in that human autopsy studies have never found pathology-specific forms of α-synuclein in the stomach or other GI location in the absence of such in the brain. Also, previous autopsy studies have focused on the GI tract itself but not on the proposed gut-to-brain conduit, the vagus nerve. It has been rightfully argued that since nervous tissue is much less concentrated in enteric walls than in CNS tissue, the apparent primacy of brain α-synuclein pathology may only be due to its much greater endowment in this respect. The vagus nerve is, like the CNS, 100%nervous tissue, and if it does serve as the connection through which α-synuclein pathology makes its passage from gut to brain, it, as well as a GI location, should be affected in at least a small percentage of normal subjects as the only manifestation of α-synuclein pathology. About 25%of clinically normal elderly subjects, dependent on age, have a limited brain distribution of α-synuclein pathology, most often in the olfactory bulb, brainstem and/or amygdala. These “incidental Lewy body disease” (ILBD) subjects also have reduced striatal dopaminergic markers [63–65], suggesting that they represent prodromal PD or dementia with Lewy bodies (DLB). If α-synuclein pathology begins in the stomach and then passes through the vagus nerve to the brain, it might be expected that for a similar percentage of normal older people, α-synuclein pathology would be limited to the stomach and/or vagus nerve, but be lacking in the CNS.
MATERIALS AND METHODS
Human subjects
The study took place at Banner Sun Health Research Institute (BSHRI), which is part of Banner Health, a non-profit healthcare provider centered in Phoenix, Arizona. BSHRI and the Mayo Clinic Arizona are the principal members of the Arizona Parkinson’s Disease Consortium. Brain necropsies and neuropathological examinations were performed on elderly subjects who had volunteered for the Arizona Study of Aging and Neurodegenerative Disorders (AZSAND)/BSHRI Brain and Body Donation Program (BBDP) [66]. Procedures involving experiments on human subjects are done in accord with the ethical standards of the BSHRI Institutional Review Boards and all subjects or their legally-authorized representatives signed a written informed consent. The majority of BBDP subjects are clinically characterized at BSHRI with annual standardized test batteries that include movement disorders and cognitive/neuropsychological components. Additionally, private medical records are requisitioned and reviewed for each subject and a postmortem questionnaire is conducted with subject contacts to help determine the presence or absence of dementia and parkinsonism for those subjects that did not have a recent standardized antemortem evaluation.
Subjects received complete neuropathological examinations while blinded to clinical diagnoses as described previously [66]. Specific consensus diagnostic criteria were used for PD, requiring 2 of 3 cardinal signs of rest tremor, rigidity or bradykinesia as well as substantia nigra α-synuclein pathology and pigmented neuron loss. Subjects with any brain α-synuclein pathology but who lacked dementia or parkinsonism were termed incidental Lewy body disease (ILBD).
Case selection was done by searching the BBDP database for all those that had died and had a full clinical evaluation and full autopsy including vagus nerve and/or stomach sampling with immunohistochemical staining for α-synuclein pathology, done with a method specific for α-synuclein phosphorylated at serine-129. Vagus nerve samples were taken adjacent to the midpoint of the carotid artery in the neck while stomach samples were taken midway along the greater curvature.
Histological methods
The process leading to the choice and evaluation of immunohistochemical methods for demonstrating pathological α-synuclein has been described in previous publications [16, 67–69]. The standard method used at AZSAND employs proteinase K pretreatment, which not only results in superior epitope exposure but also may assist with the pathological specificity of the stain by digesting normal, non-aggregated α-synuclein, which is abundant in all nervous tissue. Using an antibody specific for α-synuclein phosphorylated at serine 129 (pSyn) [70–74] also helps identify stained structures as pathological since normal control subjects do not have pSyn-immunoreactive brain tissue elements [4, 75]. Complete details of the staining procedure have been previously described [76] and so only a brief description is given here.
From each postmortem subject, three sections from vagus nerve and three sections of stomach were stained and examined. Formalin-fixed, paraffin-embedded 5–7μm sections were deparaffinized and treated with 1:100 proteinase K (Enzo Life Sciences, Farmingdale, NY) at 37°C for 20 min, followed by suppression of endogenous peroxidase activity with 1%hydrogen peroxide for 30 min, incubation in primary antibody against α-synuclein phosphorylated at serine 129, diluted 1:10,000 [71–74], incubation in biotinylated secondary antibody, avidin-biotin peroxidase complex (ABC, Vector Laboratories; Burlingame, CA) and 3,3’-diaminobenzidine (DAB; Sigma, St. Louis, MO) with saturated nickel ammonium sulfate and imidazole. All solutions subsequent to proteinase K, and all wash steps, excluding DAB incubation, were carried out in 0.1 M PBS with 0.3%Triton X-100, pH 7.4. Sections were then counterstained in 1%Neutral Red. Positive neuronal perikarya and nerve fibers are bluish-black while background and negative tissue structures are red. Cases were staged with the Unified Staging System for Lewy Body Disorders (USSLB) [3, 4].
Statistical tests
Group proportions in the three diagnostic groups were compared with chi-square tests. Group means were compared with one-way analysis of variance and post-hoc Newman-Keuls tests or two-way, unpaired t-tests.
RESULTS
Subjects included 53 clinicopathologically diagnosed with PD, 33 with ILBD and 111 who were clinically non-demented without parkinsonism and had no CNS pSyn (Table 1). The subjects were predominantly of advanced age, with the mean age ranges for the diagnostic groups falling between 83 and 87 years. Median disease duration for PD cases was 13 years, ranging from 2 to 44 years. PD cases were significantly younger (p < 0.05) than normal or ILBD cases, were more likely to be male when compared to the normal group (p = 0.006) and had higher UPDRS scores than the other groups (p < 0.0001). The mean postmortem intervals ranged between 4.1 and 4.6 h and analysis of variance showed that there were no significant differences amongst groups. Vagus nerve was available for 26 ILBD, 36 PD and 111 normal control subjects. Stomach was available for 30 ILBD, 52 PD and 102 normal control subjects. Seven control cases had a family history of PD but as the pedigrees were not strongly suggestive of autosomal dominance, none were genetically tested for PD-associated mutations or polymorphisms. Additionally, none of the ILBD cases had a PD family history but two were tested for LRRK2 (both cases) and GBA (one case) and were negative for both markers; twelve PD cases had a PD family history and eight were tested but all were negative for both LRRK2 and GBA.
Clinical characteristics of study subjects, by clinicopathological diagnosis, age, sex, last motor UPDRS score and disease duration. Means and standard deviations (SD) are given. ILBD, incidental Lewy body disease; PD, Parkinson’s disease; PMI, postmortem interval in hours; UPDRS, Unified Parkinson’s Disease Rating Scale, Part III; Dis Dur, disease duration in years. *PD cases were significantly younger (p < 0.05) than normal or ILBD cases, were more likely to be male when compared to the normal group (p < 0.05) and had higher UPDRS scores (p < 0.0001). The groups did not differ in terms of PMI
1Not all subjects had both vagus nerve and stomach available. See text for details. 2For 15 normal cases, UPDRS scores were not available. 3For 5 ILBD cases, UPDRS scores were not available. 4For PD, 16 scores were done while on medication, 35 while off medication; 2 were not done.
As in our previous investigations [1, 76–78], only staining that was morphologically consistent with nerve fibers was considered to be specific for stomach or vagus nerve α-synuclein pathology. Immunoreactive nerve fibers were present within the stomach or vagus nerve of 42/52 (81%) and 32/36 (89%) PD subjects, as compared to 5/30 (17%) and 12/26 (46%) of ILBD subjects (Table 2). No pSyn was present in stomach (102 subjects) or vagus nerve (111 subjects) of control subjects. Immunoreactive nerve fibers, when present in PD and ILBD subjects, were mostly sparse but occasionally focally frequent in the vagus nerve, and sometimes had abnormally large swollen segments, consistent with dystrophic change (Fig. 1a, b). In the stomach, immunoreactive nerve fibers were found in all layers (Fig. 1c-f) but most frequently were found in the submucosa, where they were often closely applied to the external surface of arterioles or within small nerve fascicles.
Neuropathological characteristics of Lewy body disease study subjects, by vagus and stomach pSyn status. Means and standard deviations (SD) are given. ILBD, incidental Lewy body disease; PD, Parkinson’s disease
1Missing USSLB stage and brain pSyn load for one case. 2Missing brain pSyn load in three cases.
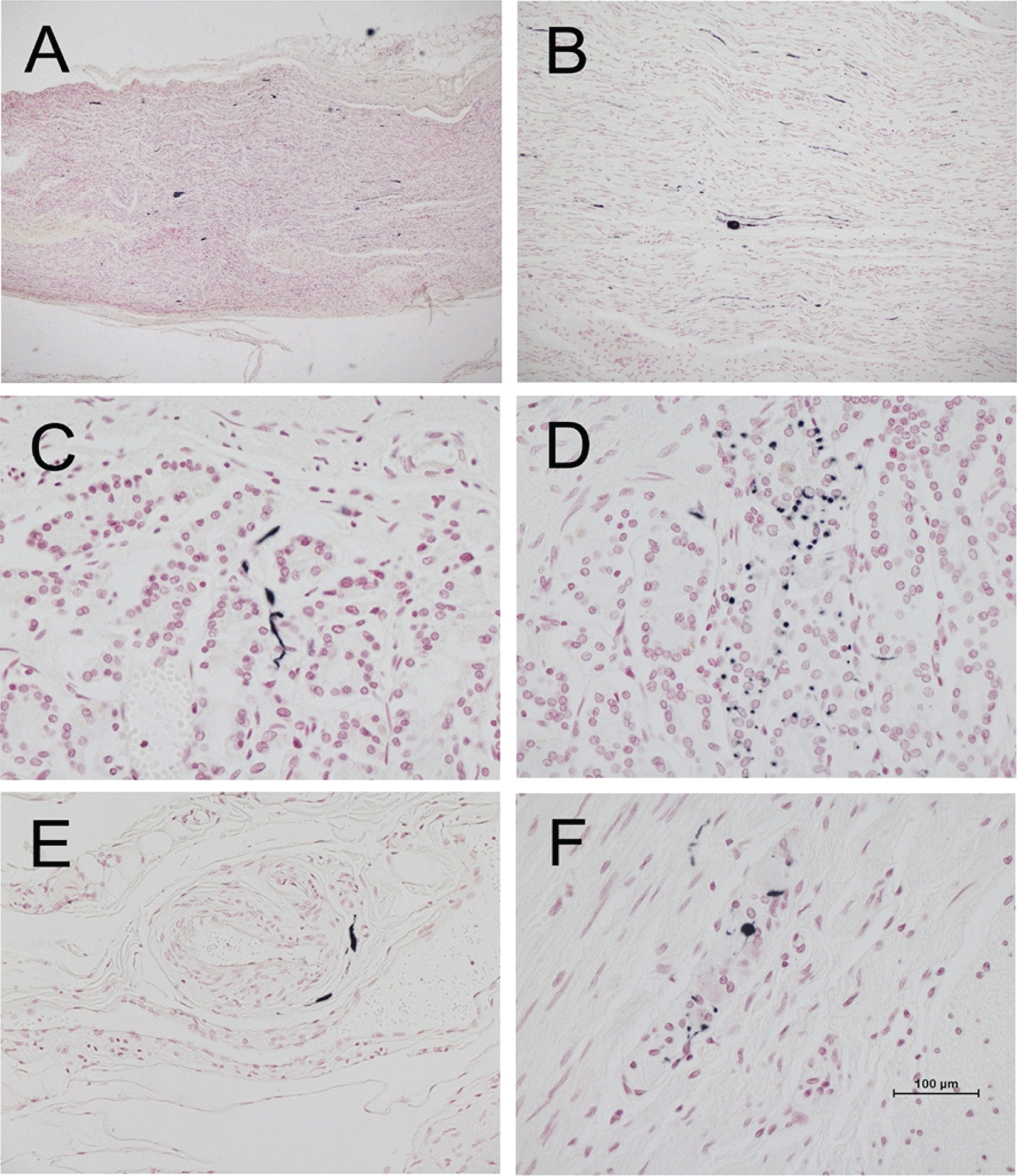
Photomicrographs of vagus nerve and stomach from four different PD subjects, immunohistochemically stained for phosphorylated α-synuclein (black) and counterstained with Neutral Red (red). A and B are longitudinal sections of vagus nerve at low (A) and medium (B) magnification. C and D show short fibers and puncta in the stomach mucosa. E shows short fibers applied to the peripheral surface on an arteriole in the submucosa. F shows puncta within an intermyenteric ganglion. Calibration bar in F represents 100μm for C-F, 400μm for B and 800μm for A.
Cases, both ILBD and PD, that were pSyn-positive in stomach or vagus tended to have higher brain Unified LB stages and higher total brain pSyn loads than those that were pSyn negative, and, on average, ILBD cases with vagus or stomach pSyn were in USSLB Stage IIa or IIb (brainstem or limbic-predominant) while PD cases were most often in Stage III (brainstem and limbic), although some ILBD cases were in Stage III and some PD cases were in Stage IV (Table 2). All cases with vagus or stomach pSyn were in Unified Stage IIa (brainstem predominant), IIb (limbic predominant) or higher, with no olfactory bulb-only cases (Stage I).
DISCUSSION
This is the first comprehensive assessment of the prevalence of stomach and vagus nerve pSyn in PD, ILBD and control subjects. The results show that stomach pSyn was present in 81%and vagus nerve pSyn was present in 89%of autopsy confirmed PD subjects, while in ILBD, pSyn was present in stomach in only 17%and in vagus in only 46%of subjects. There was no pSyn found in either the stomach or vagus nerve from any of the more than 100 clinically normal control subjects without brain pSyn. The lack of pSyn in stomach and vagus nerve, in subjects without any brain pSyn, as well as the lesser prevalences of pSyn in stomach or vagus nerve as compared to brain in all of the PD and ILBD cases investigated suggest that the spread of pSyn to the stomach and PNS occurs subsequent to the establishment of threshold brain pSyn loads. Additional evidence for this conclusion is that USSLB Stages and total brain pSyn loads were higher in ILBD and PD cases with stomach or vagus pSyn.
A critical question has been whether or not pSyn begins in the brain or within elements of the PNS [79–81]. The present results do not support the concept that the initiation of α-synuclein pathology in Lewy body disorders begins in the PNS rather than the brain. Non-motor accompaniments of PD may predate or occur early in the motor progression [49, 82–85] but the results for ILBD subjects suggest that even in these clinically prodromal subjects, pSyn has already been established in the brain. Autopsy studies of relatively small numbers of subjects with ILBD have demonstrated a high prevalence of pSyn within the spinal cord, sympathetic ganglia, adrenal medulla and upper GI tract [1, 43] but no study to date has found pSyn in the spinal cord or in PNS sites in the absence of brain involvement, with the possible exception of Fumimura et al. [45] who reported one case out of 783 with adrenal medulla as the only site with pSyn. One other case report, by Miki et al. [86] exists of Lewy body pathology restricted to the heart and stellate ganglion. As we have tested only one potential “body-to-brain” route, this leaves open the possibility that isolated peripheral synuclein pathology might exist where it is has not yet been diligently searched for. For example, synuclein pathology might occur first in the sympathetic ganglia and from there retrogradely proceed to the CNS through preganglionic axons to the intermediolateral spinal cord, or may pass to the CNS by a hematogenous route [61].
Although we used an immunohistochemical method that has been repeatedly demonstrated to be highly sensitive and specific for both CNS and PNS α-synuclein pathology, as found in multi-center blinded studies [16, 78] and biochemical studies [74], it is possible that the initial form of peripheral α-synuclein pathology may differ from that commonly seen in the CNS. Alternate forms, among many possibilities, may include non-phosphorylated α-synuclein, α-synuclein oligomers, truncated α-synuclein [87–90] and α-synuclein aggregates [39, 91]. It is possible that conversion of normal α-synuclein to pathological varieties may occur more commonly in PNS locations other than stomach, perhaps on the basis of locally high normal α-synuclein concentrations [19], or due to inflammation [92–94]. It is possible that the initial “seeding” of synucleinopathy, whether from the environment or by spontaneous internal generation, is accomplished by transiently-existing forms that may not locally incite the more familiar forms of pSyn but do so in the CNS once transmitted there. Assessment of the PNS with α-synuclein seeding assays such as RT-QuIC or protein misfolding cyclic amplification (PMCA) may be more sensitive than IHC and may be more effective at uncovering PNS α-synuclein pathology [95] although direct comparisons so far have found IHC and seeding assays roughly equivalent in gastrointestinal tissue and skin [96–98].
As mentioned, our IHC method uses a highly-characterized antibody [74] and proteinase K pretreatment to eliminate non-aggregated α-synuclein. We have repeatedly compared our IHC method with multiple other methods, in studies that have used both autopsy and clinical gold standards, studies involving multiple centers and in studies with enforced, third-party blinding [4, 99]. Our IHC method always performed amongst the best methods in these studies. We are aware of the argument that has been advanced by some researchers that antibodies to p-serine 129 α-synuclein may be less sensitive to α-synuclein pathology than antibodies to unmodified synuclein because p-synuclein may be only a minor component in α-synuclein pathological aggregates, but again, in several multicenter studies, including some with third-party blinding, our pSyn IHC method has performed similarly as the best IHC methods using antibodies to unmodified synuclein. No other putative IHC biomarker of synuclein pathology has been so extensively and rigorously tested and therefore the suggestion that other methods might be significantly more sensitive and specific than our method remain speculative.
An issue we have not been able to explore is the possibility that a body-first stage might occur as long as 20 years or more before clinical signs. Although we expect that at least some of the control subjects in our study were likely to have been in a preclinical or prodromal phase when they died, a large autopsy series of much younger subjects might better address this possibility. However, as the rates of natural death and autopsy are much lower in these younger age groups, it would be necessary to have access to a forensic autopsy series and such access has become legally restrictive.
Borghammer and Van Den Berge [100] have posited the existence of two subtypes of PD, one that is “CNS-first” and one that is “PNS-first”. Somewhat similarly, Hallett et al. have suggested that CNS and PNS synucleinopathy might proceed in parallel [101]. However, as Borghammer and Van Den Berge admit, if there is a PNS-first subtype, or if PNS and CNS synucleinopathy occur more or less in parallel, and if these subjects are more than a rare occurrence, it should still be possible to find, in a large autopsy series, at least a few cases with verified PNS α-synuclein pathology in the absence of CNS synucleinopathy, and this has not occurred in several such autopsy series including ours. It must be admitted, as Borghammer and Van Den Berge suggest, that sampling of the GI tract and/or other PNS regions has likely not yet been adequate and that PNS α-synuclein pathological species might be transported through the vagus nerve over a short time window, although for the latter scenario it would have to also be assumed that vagal transport only occurs for a short time and then ceases, despite ongoing accumulation of synuclein pathology in the gut. We would add that bilateral asymmetry of CNS and PNS α-synuclein pathology might also complicate the issue although we have investigated this and found no asymmetry of peripheral synucleinopathy, at least for the submandibular gland [102]. Borghammer and Van Den Berge also cite studies that have found a high prevalence of aggregated synuclein in the GI tract of normal subjects including children [103–105], potentially evidence for the PNS-first hypothesis, but ordinarily the finding of a disease biomarker in large numbers of normal subjects would bring into question its specificity for the disease so the relevance to PD of the aggregated synuclein found with these methods is questionable unless one proposes that additional host or environmental factors are required to convert these aggregates into truly pathogenic forms. As with other alternative hypotheses, these scenarios are extremely difficult if not impossible to absolutely disprove.
In conclusion, the results presented here are most consistent with a first appearance of phosphorylated α-synuclein pathology in the CNS, with later spread, often at a premotor stage, to the PNS.
Footnotes
ACKNOWLEDGMENTS
This study was funded by grants from the National Institute of Neurological Disorders and Stroke (U24 NS072026) the National Institute on Aging (P30 AG19610), the Arizona Department of Health Services (contract 211002), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001) and the Michael J. Fox Foundation for Parkinson’s Research.
CONFLICT OF INTEREST
The authors declare the following potential conflicts of interest:
TGB: Consultant, scientific advisory board and stock options with Vivid Genomics; Honorarium for invited lecture from Roche Diagnostics.
CHA: Consultant, Amneal Pharmaceuticals; Eisai Pharmaceuticals; Jazz Pharmaceuticals; Neurocrine, Biosciences; Cionic Inc.
LIS: None
HAS: Advisory board, Acorda Therapeutics; Jazz Pharmaceuticals.
ED-D: Clinical trials, AbbVie Inc; Biogen Inc; UCB Pharma
SHM: Consultant, CNS Ratings; Adamas Pharmaceuticals; Abbott Laboratories
AJI: None
MJG: None
JEW: None
RA: None
CMN: None
GES: None