
Abstract
Background:
Previous investigations have suggested that decreased expression of glutamate transporter-1 (GLT-1) is involved in glutamate excitotoxicity and contribute to the development of Parkinson’s disease (PD), GLT-1 is decreased in animal models of PD. GLT-1 is mainly expressed in astrocytes, and the striatum is a GLT-1-rich brain area.
Objective:
The aim was to explore the function and mechanism of astrocytic GLT-1 in PD-like changes.
Methods:
In the study, PD-like changes and their molecular mechanism in rodents were tested by a behavioral assessment, micro-positron emission tomography/computed tomography (PET/CT), western blotting, immunohistochemical and immunofluorescence staining, and high performance liquid chromatography pre-column derivatization with O-pthaldialdehida after downregulating astrocytic GLT-1 in vivo and in vitro.
Results:
In vivo, after 6 weeks of brain stereotactic injection of adeno-associated virus into the striatum, rats in the astrocytic GLT-1 knockdown group showed poorer motor performance, abnormal gait, and depression-like feature; but no olfactory disorders. The results of micro-PET/CT and western blotting indicated that the dopaminergic system was impaired in astrocytic GLT-1 knockdown rats. Similarly, tyrosine hydroxylase (TH) positive immune-staining in neurons of astrocytic GLT-1 knockdown rats showed deficit in cell count. In vitro, knockdown of astrocytic GLT-1 via RNA interference led to morphological injury of TH-positive neurons, which may be related to the abnormal calcium signal induced by glutamate accumulation after GLT-1 knockdown. Furthermore, the GLT-1 agonist ceftriaxone showed a protective effect on TH-positive neuron impairment.
Conclusion:
The present findings may shed new light in the future prevention and treatment of PD based on blocking glutamate excitotoxicity.
INTRODUCTION
Parkinson’s disease (PD) is a progressive degenerative disease of the central nervous system, which mainly manifests as cardinal movement disorders, including hypokinesia, bradykinesia, muscle rigidity, limb tremor and abnormal posture and gait [1]. Pathologically, progressive loss of dopaminergic neurons in the substantia nigra and formation of Lewy bodies are the main features. PD severely jeopardizes the health of middle-aged and elderly individuals and deprives them of working and living capacity [2]. With increased aging of the population, the number of PD patients is also on the rise [3].
Although it has been more than 200 years since PD was first recognized, and more and more in-depth studies have been carried out, the etiology and pathogenesis of PD are still unclear. At present, it is generally accepted that the morbidity of PD is closely related to age, environmental, and genetic factors [4]. In recent years, with the establishment and application of animal and cell models, researchers have found that mitochondrial dysfunction, oxidative stress, neuroinflammation and excitatory amino acid toxicity caused by many factors may be the key pathophysiological processes leading to the injury, degeneration, death, and loss of dopaminergic neurons [5]. Of these processes, the toxic effect of excitatory amino acids have become a research hotspot among the etiological hypotheses of PD [6].
Glutamate, one of the essential and most critical excitatory neurotransmitters in the central nervous system, has naturally become the focus of research in the context of the excitotoxicity hypothesis. Many studies have confirmed that the disorder of glutamatergic transmission in relevant brain regions plays an important role in the development of PD [7]. The most direct evidence was from the study by Ferrarese et al. [8] in which high performance liquid chromatography (HPLC) was used to determine the level of glutamate in peripheral blood samples from 34 PD patients (PD group) and 21 age-matched healthy volunteers (control group) to indirectly monitor platelet glutamate uptake function. The results showed that glutamate uptake in the PD group decreased by more than 50%compared with the control group, and the decrease was greater in more severe PD cases. Similar findings in basic research were reported even earlier. In 1997, Hazell et al. [9] found that Parkinsonian-like changes induced by the neurotoxin MPTP might be related to reduced glutamate uptake. Therefore, glutamate uptake disorder is an important pathogeny of PD. This can be explained at the pathophysiological level as follows: the decrease in glutamate uptake inevitably leads to the accumulation of extracellular glutamate, which constantly stimulates the glutamate receptor on the cell membrane of dopaminergic neurons, thus activating intracellular second messengers such as inositol triphosphate, inducing intracellular Ca2 + overload and a downstream cascade reaction, eventually leading to the death of dopaminergic neurons [10]. This damage is accompanied by increased energy consumption and the formation of free radicals, which constitute the complete excitatory neurotoxic effect of glutamate [11].
How does glutamate uptake disorder occur in PD patients? Previous studies have suggested that as no extracellular enzyme can metabolize glutamate, the only way for cells to quickly remove extracellular glutamate is to ingest and recycle it. During this process, the glutamate-glutamine cycle between astrocytes and neurons (including presynaptic and postsynaptic neurons) is the most important physiological step [12]. This step is controlled by the key protein glutamate transporter-1 (GLT-1), which accounts for more than 90%of the total glutamate uptake in the brain. Recent studies further found reduced expression of GLT-1 in PD animal models established by induction with 6-OHDA [13], MPTP [14], and MPTP metabolite 1-methyl-4-phenylpyridinium (MPP+) [15], accompanied by a decrease in glutamate uptake. Therefore, researchers realized that the glutamate uptake disorder in PD is closely related to the decrease in GLT-1, and proposed that GLT-1 plays an important role in the development of PD.
GLT-1 is mainly expressed on astrocytes [16], and the striatum is one of the brain regions where it is abundantly expressed [17]. We observed decreased GLT-1 expression in the striatum of PD animal models at the beginning of our study, which was the same phenomenon observed in their previous studies. Accordingly, this will add new evidence to the involvement of GLT-1 in the development of PD, if we can prove that the reduction in GLT-1 in the striatum leads to Parkinsonian-like phenotype. In the present study, we downregulated GLT-1 in striatal astrocytes by Adeno-associated virus (AAV)-mediated interference in vivo and verified Parkinsonian-like phenotypic changes using animal behavior, neuroimaging, and histochemistry analyses. We also investigated the effect of GLT-1 knockdown in astrocytes on dopaminergic neurons and its possible mechanism by primary cell culture in vitro.
MATERIALS AND METHODS
Animals
Adult male Sprague-Dawley (SD) rats (weight 200–220 g) were included for the in vivo GLT-1 knockdown or the 6-OHDA-induced PD model, and adult male C57BL/6 mice (weight 25–28 g) were included for the MPTP-induced PD model. These animals were purchased from Slaccas Laboratory (Shanghai, China). All experimental mice were specific pathogen-free. Five animals in each cage were kept in the following environment-controlled rooms: temperature (21±2°C), relative humidity (55±5%), well-ventilated, light/dark ratio 12/12 h, with free access to water and food. Animal experiments were conducted in strict accordance with the Principles of Laboratory Animal Care and Use and were approved by the Institutional Animal Care and Use Committee of Soochow University (Suzhou, China).
Animal models
PD rat model induced by stereotactic injection of 6-OHDA into bilateral substantia nigra pars compacta (SNpc)
The 6-OHDA-induced PD rat model was established as previously described [18]: Rats were deeply anesthetized with 2%isoflurane inhalation and placed in a Stoelting stereotaxic apparatus (Stoelting Co., Kiel, WI, USA). According to a rat brain atlas, 6-OHDA (Sigma, St. Louis, MO, USA), which was dissolved in saline, was injected into the bilateral SNpc (8μg/4μL on one side) at a rate of 0.5μL/min using a 10 μL Hamilton syringe. The coordinates for the bilateral SNpc were –5.3 mm anteroposterior, ±1.8 mm mediolateral, and –7.8 mm dorsoventral (from the dura) from the bregma. The syringe was left in the SNpc for 4 min and then slowly retracted. Control rats underwent the same procedure, only the injectant was changed to an equal volumes of saline. Rats were tested and examined three weeks after modelling.
Subacute PD mouse model induced by intraperitoneal injection of MPTP
The MPTP-induced subacute PD mouse model was established as previously described [14, 19]. Experimental mice received an intraperitoneal (i.p.) injection of 30 mg of MPTP-HCl/kg for five consecutive days. Control mice received an i.p. injection of an equal volumes of vehicle. The mice were tested and examined on day 3 after injection.
GLT-1-knockdown rat model in unilateral striatal astrocytes
The AAV-mediated GLT-1 knockout method was performed according to the method of Zhang et al. [20]. AAV that specifically transduces astrocytes was constructed by Wuhan Institutes of Advanced Technology, Chinese Academy of Sciences with the following sequences: rAAV9-hGFAP-EGFP-5′miR-30a-shRNA (scramble)-3′miR-30a for the control group and rAAV9-hGFAP-EGFP-5′miR-30a-shRNA (GLT-1)-3′miR-30a for the knockdown group. The AAV was delivered to unilateral striatal astrocytes of SD rats by stereotactic injection with two points in the right striatum: point 1 at 0.7 mm anterior to the bregma, 2.6 mm right to the midline, and 5.0 mm subdural; point 2 at 0.3 mm posterior to the bregma, 3.6 mm right to the midline, and 5.0 mm subdural. At each point 2.3μL of 6.21×1012 vg/mL scramble-Ctrl-AAV or 2.5μL of 5.78×1012 vg/mL GLT-1-KD-AAV was injected. Some of these rats were treated with the GLT-1 agonist ceftriaxone (200 mg/kg, i.p.) once a day for 7 days from 3 days before injection of scramble-Ctrl-AAV or GLT-1-KD-AAV. Therefore, four groups were included in this part of the study, namely the Ctrl, Ceftriaxone, GLT-1 KD and GLT-1 KD + Ceftriaxone. The rats were tested and examined six weeks after injection.
Animal behavior evaluation
The rotarod test was performed in all three animal models, the sucrose preference test was carried out in the two rat models and the tail suspension test was only performed on MPTP–treated mice; the remaining behavioral tests were only carried out on GLT-1-knockdown rats.
Rotarod test
Impairment of motor balance ability is an important clinical feature of PD. The rotarod test is a common method used to evaluate the motor balance of mice, and abnormal test results are regarded as a behavioral indicator of the successful modelling of PD. Based on our previous experience [18, 21] and past studies which used the rotarod test to evaluate the characteristic behavior of a unilateral traumatic PD model [22, 23], we carried out the rotating rod test not only in the 6-OHDA-induced rat model and MPTP-induced mouse model, but also in rats with unilateral knockdown of GLT-1. The test was performed using the Rotarod system (ZH-300, Zhenghua, Anhui Province, China) to test forced motor function and balance. After three consecutive days training (three times per day), experimental animals were tested three times (the interval between adjacent tests was 30 min) in the formal test and the rotational speed was increased to 15 rpm/min (for mice) or 25 rpm/min (for rats) in a test session, and the time on the rod during the 300s-test was recorded and the average time was taken as the final result.
Tail suspension test
Previous studies, especially pharmacological studies, found that normal mice do not like being hung-up by the tail and will actively struggle in this state, whereas depressed mice will give up struggling and show despair. Therefore, the tail suspension test is often used to evaluate depression-like behavior in mice. The tail suspension test was performed as previously described [24]: Briefly, the mice were hung up by their tails 50 cm away from the table using medical adhesive tape 30 mm from the start of the tail and the tape was attached to the iron cross bar. To prevent tail climbing behaviors, passing mouse tails were placed inside through a small plastic cylinder prior to suspension. Mouse behaviors was recorded over 6 min using a video camera. A person was trained to measure out the immobility time in mice in the last 4 min of the 6-min video recording.
Sucrose preference test
The sucrose preference test is an important method to evaluate the depression-like behavior core phenotype-pleasure deficiency of animals, especially in rats, which are un-suitable for the tail suspension test. The sucrose preference test was performed as follows: Briefly, rats were trained to drink 1%sucrose solution (w/v) from two bottles for 1 day. On the 2nd day, one of the bottles was replaced with a water bottle. After adaptation, the rats were deprived of water and food for 12 h followed by the sucrose preference test. The rats were presented with two different bottles for 12 h: one filled with 100 mL of water and the other with 100 mL of 1%sucrose solution (w/v). During the test, the positions of the two bottles (right/left) were randomly placed and then reversed after 6 h. One rat per cage was include for this test. According to our previous report [25], the outcome measures were the weight-adjusted intake of water or 1%sucrose (i.e., mL/kg) and the percent of sucrose preference [i.e., 100 sucrose solution intake (mL) / total fluid intake (water plus sucrose solution) (mL)].
Buried food pellet test
The Buried food pellet test is a method for evaluating olfactory function in animal studies, and its calculated indicator is the time the animal takes to find buried a food pellet under bedding. In this study, the buried food test was performed as previously described [26]: in brief, rats were placed in a clean cage (40 cm length×30 cm width×20 cm height) and exposed to the pellet for 2–3 consecutive days before the test. The rats were deprived of food for 18–24 h and habituated to the testing room for 1 h. Then a subject rat was placed in a clean cage containing clean bedding 3 cm deep for testing. Following 1 h of habituation, one pellet was placed 0.5 cm below the bedding. The test rat was removed from its home cage, placed in the center of the test cage, and the timer was started. The trial was ended when the pellet was found, and the time taken for in this process was recorded. If the rat did not find the pellet within 5 min, a time of 300 s was noted. We measured the latency time of each rat in five tests and the average of these tests was considered the final outcome.
Open field test
In the open field test, the total distance (m) of autonomous movement by each animal within a specified time is used to evaluate the locomotion of experimental animals [27]. Each rat was tested separately. The rat was placed in the center of a box (80×80×40 cm) connected to a camera. The color of the inner and bottom side of the behavior box was black (contrary to the color of the rats). The locomotor tracks were continuously recorded by the automated Flex field/Open Field Activity System (ANY-maze, Stoelting, USA). Each rat was recorded for 600 s and repeatedly recorded three times at intervals of 30 min. The experimental data were automatically transmitted to the computer for further analysis. The whole experiment was conducted in a quiet room with 80 lux light, and each rat was not subjected to external stress for at least 12 h before the test, and the inner wall and bottom of the box was cleaned with 5%ethanol after each test, to prevent the remaining information from the previous rat (such as, urine and odor) affecting the next test results.
Gait analysis
Abnormal gait is a characteristic manifestation of PD patients. The traditional ink footprint evaluation method of animal gait generates limited data, and the analysis is too subjective. In this study, the TreadScan gait analysis system (CleverSys Inc, Reston, VA, USA) was used for gait analysis [28]. The system includes an enclosed lucent walkway with light from a white fluorescent tube, and a video camera was connected to monitor the run positioned underneath the platform. Rats crossing the walkway were monitored by a high-speed color camera connected to a computer with customized image acquisition program. Before the test, each rat was trained in adaptability for three days (twice a day for 10 min at a speed of 20 m/min). During training, if the rat stopped walking or turned around, we manually relocated it until the whole process was completed. In the test, each rat was placed on the walkway at a speed of 20 m/min and repeatedly performed 3 uninterrupted runs. Each rat was videotaped for 20 s each time. Treadscan 4.0 software was used for data analysis. Firstly, the foot model data through a single frame were built, then using manual automatic preview the effective image was segmented, and finally, the gait parameters were analyzed. A double blind method was used during the test, which was carried out by two trained experimental personnel.
Cell culture
Culture and treatment of primary astrocytes
Primary astrocytes were derived from the cortex of 1–3-day-old SD rats and were maintained in Dulbecco’s modified eagle’s medium/F12 (Gibco, Grand Island, USA) supplemented with 10%fetal calf serum (Gibco, Grand Island, USA). Microglia were depleted by treatment with 1.5 mM L-leucine methyl ester (Changshu, Suzhou, Jiangsu, China) on day 9–14. Astrocytes were equilibrated in humidified air containing 5%CO2 at 37°C. Transfection of siRNA in astrocytes was performed on day 17–18 using riboFECTTM CP Reagent (RioBio, Guangzhou, China) following the manufacturer’s instructions. The siRNA sequence targeting GLT-1 was used as follow: 5′-GCTCTCACTGACTGTGTTT-3′. The GLT-1-knockdown astrocytes were harvested or co-cultured with primary mesencephalic neurons 72-h post transfection.
Culture and treatment of primary mesencephalic neurons
Primary mesencephalic neurons containing 5%dopaminergic neurons (Supplementary Figure 1) were harvested from embryonic day 12–14 SD rats. The midbrain was quickly detached on the cold stage. The mesencephalic tissues were digested by 0.125%trypsin for 15 min at 37°C. After centrifugation, the cells were re-suspended in neurobasal medium, passed through a filter, and plated on poly-L-lysine-coated dishes with neurobasal medium (Gibco, Grand Island, USA) plus 1%glutaMAX and 2%B-27. On day 2, the culture medium was changed to complete medium and 1μM cytosine arabinoside (Ara-C). On day 4, the culture medium was changed to the above medium without Ara-C, and half of the medium was changed every 72 h thereafter until the mesencephalic neurons matured on approximately day 10–11, which were then co-cultured with GLT-1-knockdown astrocytes in transwell system.
Co-culture of primary astrocytes-primary mesencephalic neurons
GLT-1-knockdown or Ctrl primary astrocytes were co-cultured with primary mesencephalic neurons in the transwell co-culture system with astrocytes in the upper chamber and mesencephalic neurons in the lower chamber. Ceftriaxone was simultaneously added to some of the co-cultured cells at a final concentration of 1 mM for 72 h. Thus four groups were included in this part of the study, namely the negative control (NC), Ceftriaxone, GLT-1 KD and GLT-1 KD + Ceftriaxone. Cells were co-cultured in neurobasal medium containing 2%B-27, 1%glutaMAX, and 1%Penicillin-Streptomycin for 72 hr. After that the supernatant was collected to measure the concentration of glutamate.
Micro-positron emission tomography/computed tomography (PET/CT) imaging assay
An Inveon small animal PET scanner (Siemens Preclinical Solutions, used by Jiangsu Huayi Technology Co., Ltd., Changshu, Suzhou, Jiangsu, China) was utilized to evaluate changes in the dopamine system in GLT-1 knockdown SD rats. The rats were anesthetized with 2%isoflurane inhalation. A total volume of 11.12 MBq (300μCi) 18F-FPCIT, a dopamine transporter (DAT) PET radiotracer with high specificity and favorable in vivo imaging properties, was injected into the tail vein of rat. Image acquisition was performed 30 min after tail vein injection of 18F-FPCIT. PET was performed initially with the following parameters: energy peak, 511 keV; window width, 3.5 E0 SUV-bw; resolution, 0.775 mm/pixel; and matrix, 128×128. Each rat required a 10-min acquisition; CT was performed after PET scanning in the same bed position using the following parameters: frame resolution, 256×512; tube voltage, 80 kVp; current, 0.5 mA; and exposure time, 500 ms/frame. 18F-FPCIT was provided by Jiangsu Huayi Technology Co., Ltd., Changshu, Suzhou, Jiangsu, China.
Measurement of glutamate in supernatant by high performance liquid chromatography (HPLC) pre-column derivatization with O-pthaldialdehida (OPA)
Glutamate in the cell culture supernatant was measured by HPLC-pre-column derivatization with the OPA system (AB Sciex, USA), and the help of the Institute of Neuroscience, Chinese Academy of Sciences, Shanghai, China.
Western blotting
Striatal tissues were dissected from the brain of experimental mice and homogenized in ice-cold stringent RIPA buffer. Homogenates were centrifuged at 12,000 rpm for 20 min at 4°C. The protein concentration was measured by BCA Protein Assay Kit (Thermo Scientific, USA). Cell lysates were prepared with lysis buffer containing 150 mM NaCl, 25 mM Tris, 5 mM EDTA, 1%Nonidet P-40, pH 7.5, with protease inhibitor (Roche Diagnostics, Germany) and phosphatase inhibitor (Thermo Scientific, USA) cocktail tablets. Samples were separated on 8–12%SDS-PAGE and transferred onto polyvinylidene fluoride membranes. This was followed by blocking with 10%non-fat powdered milk in 0.1%Tris-buffered saline with 0.05%Tween-20 (TBST) at room temperature for 1 h. Samples were incubated overnight at 4°C with the following primary antibodies: anti-GLT-1 (ab41621, 1 : 800; Abcam, Cambridge, MA, USA); anti-tyrosine hydroxylase (TH) (T1299, 1 : 2500; Sigma–Aldrich, St. Louis, MO, USA); anti-GFAP (ab7260, 1 : 10000; Abcam Cambridge, MA, USA); anti-DAT (ab128848, 1 : 1000; Abcam Cambridge, MA, USA); anti-Phospho-CaMKII (Thr286) (12716, 1 : 1000; Cell Signaling Technology, Beverly, MA, USA); and anti-β-actin (A3854, 1 : 5000; Sigma–Aldrich, St. Louis, MO, USA). The integrated intensity of the gray value of blots was quantified by Image J software (National Institutes of Health, Bethesda, MD, USA).
Immunohistochemistry and immunofluorescence of brain sections
The brain fixed in 4%paraformaldehyde was dehydrated with a series of 15%and 30%sucrose solution at 4°C. Brain sections 30μm thick containing the striatum and SNpc were cut using a cryostat (Leica, Wetzlar, Germany). Brain sections were washed in 0.01 M phosphate-buffered saline (PBS) and blocked in 5%bovine serum albumin containing 0.1%Triton X-100 for 1 h and incubated with the anti-TH antibody (T1299, 1 : 1000; Sigma-Aldrich, USA) at 4°C overnight. The sections were washed in 0.01 M PBS three times and incubated with HRP-conjugated secondary antibodies. Then the sections were stained using the DAB kit (GK500705, Genentech, China). After visualization with a Zeiss microscope (AXIO SCOPE A1, Zeiss Corp, Germany), five brain slices with the same anatomical structure from each of three rats from each group were selected. The number of TH-positive neurons in bilateral SNpc were blind-counted in one out of every six neighboring sections. In order to better show the range of striatum and the specificity of AAV transducing astrocytes, we performed immunofluorescence staining on some brain slices in the GLT-1 knockdown experiment. The procedures were basically the same as immunohistochemical staining, but the primary antibody and secondary antibody were different, as follows: DARPP32 (ab40801,1 : 200; Abcam, Cambridge, MA, USA), GFAP (ab7260, 1 : 1000; Abcam, Cambridge, MA, USA); Alexa FluorTM 555 goat anti- rabbit IgG (H + L) (A21429, 1 : 1000; Invitrogen, USA); Iba1 (ab5076, 1 : 1000; Abcam, Cambridge, MA, USA); Alexa FluorTM 488 goat anti-goat IgG (H + L) (a11056; 1 : 1000; Invitrogen, USA).
Immunofluorescence of primary cells
Primary astrocytes or primary mesencephalic neurons were cultured on coverslips, which were washed in PBS and fixed with 4%paraformaldehyde for 10 min. Cells were blocked in 5%BSA with 0.1%Triton X-100 for 1 h and incubated with primary antibodies overnight at 4°C, rinsed with PBS, and incubated with Alexa Fluor 488-conjugated goat anti-mouse and Alexa Fluor 594-conjugated goat antirabbit IgG for 2 h at room temperature. After staining cellular nuclei with 4′, 6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA, USA), the cells were visualized with a Zeiss confocal microscope (LSM700, Carl Zeiss, Germany).
Sholl analysis
We performed Sholl analysis of the branch of TH positive neurons based on our experience in studying microglia [29]. Briefly, images immunostained with TH antibody of primary mesencephalic neurons on coverslips were captured. The maximal projection image of the TH signal in the stratum radiatum was adopted for analysis. The plugin of Sholl analysis applied in ImageJ automatically draws serial concentric circles at 10-μm intervals from the center of the DAPI signal to the end of the most distant process in each single TH positive neuron and analyzes the number of intercepts of TH processes in each circle and the ramification index.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 8. Numeric data that conformed to a Gaussian distribution were displayed as mean±standard error of the mean (SEM). All statistical analyze were based on the results from ≥3 independent biological samples and were repeated on different experimental days. Numeric data were compared with two-tail t-test, two-way ANOVA or three-way ANOVA. p value < 0.05 was considered statistically significant.
RESULTS
Striatal GLT-1 was reduced in PD murine models
Currently there are no reports on the change of GLT-1 level in the striatum of PD patients and previous preclinical studies have reported limited but inconsistent results of striatal GLT-1 in animal models of PD. It is generally believed that GLT-1 is decreased in the striatum in animal PD models. In this study, we detected GLT-1 expression in the striatum of a subacute PD mouse model induced by i.p. of MPTP and a PD rat model induced by stereotactic injection of 6-OHDA into bilateral SNpc. We confirmed successful modeling of PD in MPTP-treated mice by observing the loss of TH-positive neurons in the SNpc (Fig. 1A, t = 9.470, df = 6, p < 0.0001), reduced TH protein (t = 10.50, df = 4, p = 0.0005) in the striatum (Fig. 1B) and reduced on-rod time in the rotarod test (Fig. 1C, t = 5.008, df = 11, p = 0.0004). On the basis of successful modeling, GLT-1 (t = 5.683, df = 4, p = 0.0047) was found to be reduced in the striatum of MPTP-treated mice (Fig. 1B). The modeling of PD in rats induced by stereotactic injection of 6-OHDA into bilateral SNpc was also successful. There loss of TH-positive neurons was observed in the SNpc (Fig. 1E, t = 4.990, df = 6, p = 0.0025), reduced TH protein was reduced (t = 8.840, df = 4, p = 0.0009) in the striatum of 6-OHDA-rats (Fig. 1F) and on-rod time in the rotar rod test was reduced in 6-OHDA-treated rats (Fig. 1G, t = 5.771, df = 13, p < 0.0001). As expected, we found that GLT-1 (t = 4.268, df = 4, p = 0.0130) was also reduced in the stratum of 6-OHDA-treated rats (Fig. 1F). Surprisingly, there was an increase in immobility time in MPTP-treated mice in the tail suspension test (Fig. 1D, t = 3.731, df = 11, p = 0.0033) and a decline in sucrose intake in 6-OHDA-treated rats in the sucrose preference test. (Fig. 1H, t = 4.975, df = 12, p = 0.0145), suggesting possible concomitant depression-like symptoms and PD. These results suggested that GLT-1 is downregulated in the striatum of PD animal models, and this decline might occur in cases of PD associated with depression.
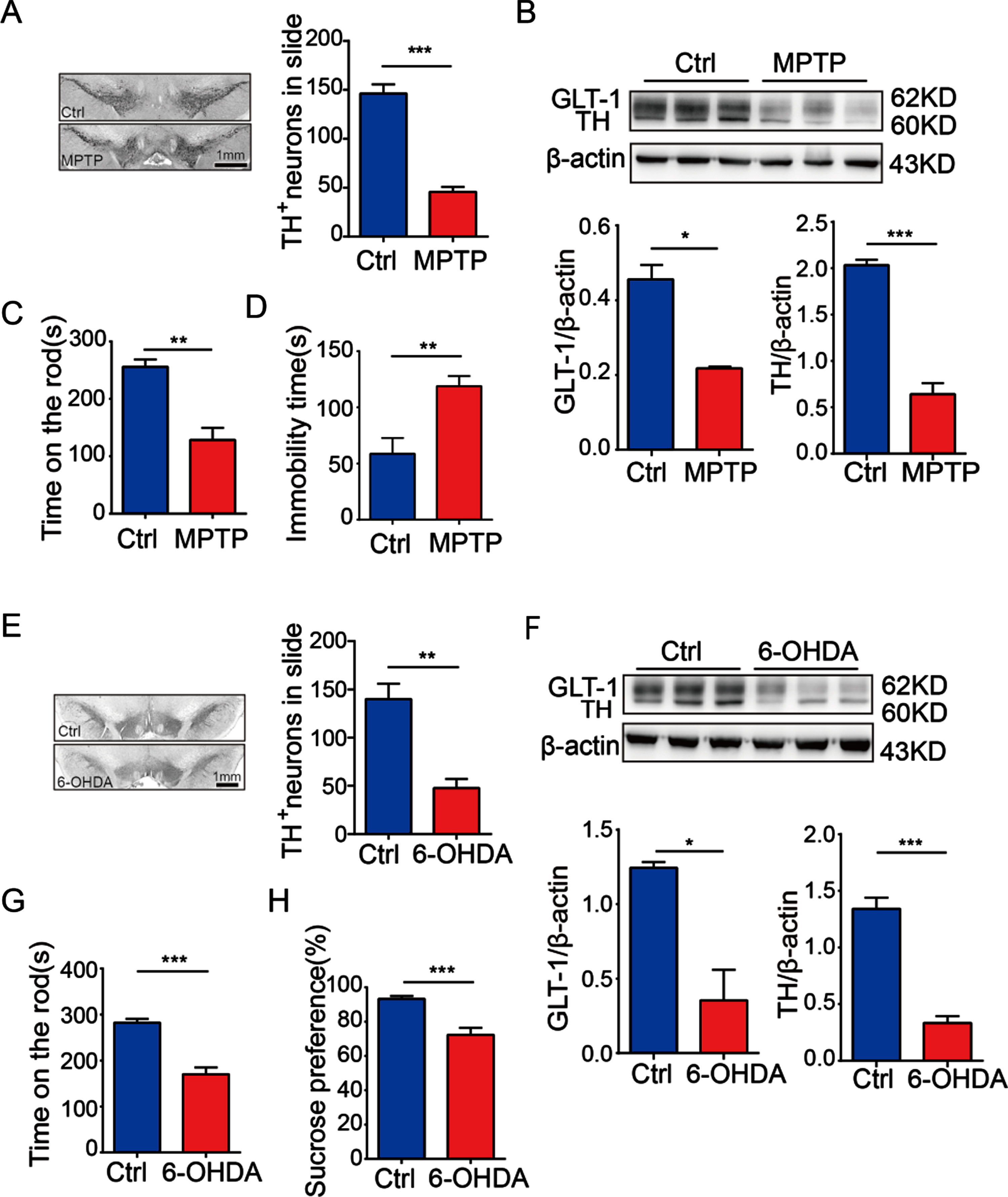
Changes in striatal GLT-1 in different PD animal models. A) Immunohistochemistry showed the changes in TH-positive neurons in the SNpc of rats in the Ctrl group and MPTP-treated group (scale bar = 1 mm). B) Western blotting was performed to detect the expression of TH and GLT-1in the striatum of subacute PD-mice induced by i.p. MPTP. C) The rotarod test evaluated motor ability of subacute PD-mice induced by i.p. MPTP. D) The tail suspension test assessed depression-like behavior of subacute PD-mice induced by i.p. MPTP. E) Immunohistochemistry showed the changes in TH-positive neuronsin the SNpc of rats in the Ctrl group and 6-OHDA group (scale bar = 1 mm). F) Western blotting was preformed to detect the expression of TH and GLT-1 in the striatum of PD-rats induced by injection of 6-OHDA into bilateral SNpc. G) The rotarod test evaluate motor ability of PD-rats induced by injection of 6-OHDA into bilateral SNpc. H) The sucrose preference test assessed depression-like behavior of PD-rats induced by injection of 6-OHDA into bilateral SNpc. Bar value = mean±SEM, n = 3 for western blotting, n = 6-9 for the behavioral study. *p < 0.05; **p < 0.01; ***p < 0.001.
Knockdown of astrocytic GLT-1 in the right striatum caused PD-like behavior in SD rats, and the GLT-1 agonist prevented this behavior
This study verified that striatum GLT-1 was indeed reduced in the 6-OHDA- and MPTP-induced PD models. Does direct knockdown of GLT-1 in the striatum lead to Parkinsonian-like changes? As GLT-1 is mainly expressed in astrocytes, we used AAV that can specifically transduce astrocytes for knockdown of GLT-1 expression in right striatal astrocytes in vivo. Three weeks after AAV transduction, rat brain slices were prepared, and co-localization of eGFP from the AAV and the astrocyte marker GFAP were observed in astrocytes by immunofluorescence staining (Fig. 2A), which confirmed the specificity of AAV for astrocytes. More importantly, it was not found to spread to other brain areas outside the striatum except the pathway through which the injection needle passed. Furthermore, the efficiency of the knockdown of astrocytic GLT-1 was examined six weeks post transduction, which showed a visible reduction in GLT-1 (three-way ANOVA: F knock x side x drug (1, 1) = 7.539, p = 0.0144; F knock x side (1, 1) = 17.38, p = 0.0007; F knock x drug (1, 1) = 6.747, p = 0.0194; F side x drug (1, 1) = 9.904, p = 0.0062; F knock (1, 1) = 0.3057, p = 0.5880; F side (1, 1) = 25.14, p = 0.0001; F drug (1, 1) = 11.07, p = 0.0043) level while astrocytes marker GFAP (three-way ANOVA: F knock x side x drug (1, 1) = 12.36, p = 0.0018; F knock x side (1, 1) = 3.036, p = 0.0942; F knock x drug (1, 1) = 19.36, p = 0.0002; F side x drug (1, 1) = 13.33, p = 0.0013; F knock (1, 1) = 269, p < 0.0001; F side (1, 1) = 5.694, p = 0.0253; F drug (1, 1) = 3.835, p = 0.0619) was increased at this time (Fig. 2B). We were then interested in the behavioral changes in animals after GLT-1 knockdown. In the rotarod test, we found that the on-rod time in the GLT-1 knockdown (GLT-1 KD group) rats was shorter than that in control rats (Ctrl group) at six weeks after GLT-1 knockdown, but treatment with ceftriaxone, an agonist of GLT-1, prevented this reduction (Fig. 2C, two-way ANOVA: F interaction (1, 28) = 0.7505, p = 0.3937; F knock (1, 28) = 11.73, p = 0.0019; F drug (1, 28) = 54.66, p < 0.0001). In the open field test, the total distance of locomotion in the GLT-1 KD group was less than that in the Ctrl group, and ceftriaxone prevented this reduction (Fig. 2D, two-way ANOVA: F interaction (1, 28) = 4.793, p = 0.0371; F knock (1, 28) = 16.96, p = 0.0003; F drug (1, 28) = 162.8, p < 0.0001). Furthermore, gait analysis showed that the average standing time (an indication of frozen gait in PD patients) (two-way ANOVA: F interaction (1, 28) = 5.703, p = 0.0239; F knock (1, 28) = 8.492, p = 0.0069; F drug (1, 28) = 46.38, p < 0.0001) and average braking time (an indication of difficulty in stopping in PD cases) (two-way ANOVA: F interaction (1, 28) = 1.052, p = 0.3138; F knock (1, 28) = 10.57, p = 0.0030; F drug (1, 28) = 71.64, p < 0.0001) of the rear right limb in GLT-1 KD rats were both longer than those in the Ctrl group, and ceftriaxone prevented these increases (Fig. 2E). Besides motor dysfunction, we then investigated whether there were changes in non-motor features of these animals six weeks after GLT-1 knockdown. In the sucrose preference test, the consumption of sucrose solution (two-way ANOVA: F interaction (1, 28) = 4.670, p = 0.0394; F knock (1, 28) = 5.759, p = 0.0233; F drug (1, 28) = 76.24, p < 0.0001) in the GLT-1 KD group was reduced compared with the Ctrl group within the same feeding period. This suggested that GLT-1 knockdown in unilateral striatal astrocytes in vivo induced a lack of pleasure in rats, which is the core phenotype of depression-like behavior. Surprisingly, ceftriaxone also prevented the reduction of sucrose consumption (Fig. 2F). However, in the buried food pellet test, we found that there was no significant difference in the time to find buried food pellets (two-way ANOVA: F interaction (1, 28) = 0.02396, p = 0.8781; F knock (1, 28) = 0.7755, p = 0.3860; F drug (1, 28) = 2.862, p = 0.1018), between the GLT-1 KD group and the Ctrl group (Fig. 2G), suggesting that olfactory function of rats after GLT-1 knockdown in unilateral striatal astrocytes was unaffected at the set observation time point.
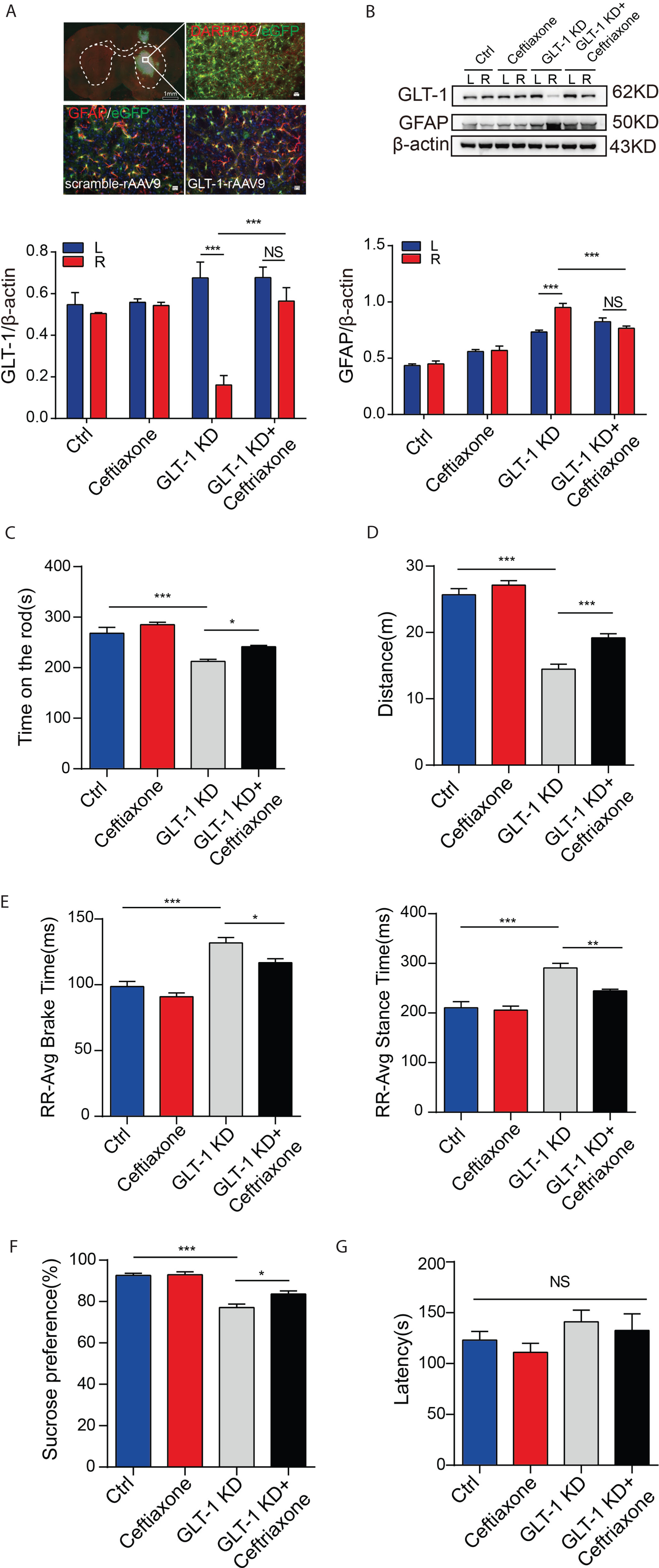
Changes in behavioral features of SD rats six weeks after knockdown of GLT-1 in right striatal astrocytes in vivo. A) Co-localization of eGFP from AAV and the astrocyte marker GFAP in rat striatal brain slices by immunofluorescence staining three weeks after AAV transduction which confirmed the specific transduction of AAV to astrocytes (scale bar = 1mm and 20.0μm). B) Western blotting showing the knockdown of GLT-1 and GFAP proliferation in the striatum six weeks after AAV injection. C) Comparison of the on-rod time in the rotarod test among the Ctrl, Ceftriaxone, GLT-1 KD and GLT-1 KD+Ceftriaxone groups. D) Comparison of the total distance of locomotion in the open field test among the Ctrl, Ceftriaxone, GLT-1 KD and GLT-1 KD + Ceftriaxone groups. E) Comparison of the average standing or braking time of the rear right limb of the rats in the Ctrl, Ceftriaxone, GLT-1 KD and GLT-1 KD+Ceftriaxone groups within the same period of time by automatic gait analysis. F) Comparison of sucrose consumption among the Ctrl, Ceftriaxone, GLT-1 KD and GLT-1 KD+Ceftriaxone groups within the same period of time. G) Comparison of the time required for rats in the Ctrl, Ceftriaxone, GLT-1 KD and GLT-1 KD + Ceftriaxone groups to find buried food pellets within the same period of time. Bar value = mean±SEM, n = 3 for immunofluorescence and western blotting, n = 8 for the behavioral study. *p < 0.05; **p < 0.01; ***p < 0.001; NS, non-significant.
Knockdown of astrocytic GLT-1 in the striatum provoked abnormal changes of the dopamine system in the substantia nigra-striatum pathway in SD rats, and the GLT-1 agonist blocked these changes
The above animal behavior studies showed that knockdown of GLT-1 in right striatal astrocytes in SD rats in vivo can lead to motor dysfunction (poor motor balance, decreased locomotor ability and abnormal gait) and non-motor symptoms (depression-like behavior). We next explored the histochemical and neuroimaging changes underlying these behavioral changes. We focused on alteration of the dopamine system in the substantia nigra-striatum pathway. Micro-PET/CT, western blotting, and immunohistochemistry were performed in the Ctrl, Ceftriaxone, GLT-1 KD and GLT-1 KD + Ceftriaxone groups 6 weeks after AAV injection. The result showed that the uptake of DAT tracer 18F-FPCIT in the GLT-1-knockdown side of the striatum was lower than that in the non-rAAV-injected side and in the Ctrl group (Fig. 3A). Moreover, the western blotting results of striatal DAT (three-way ANOVA: F knock x side x drug (1, 1) = 0.00918, p = 0.9249; F knock x side (1, 1) = 45.05, p < 0.0001; F knock x drug (1, 1) = 12.38, p = 0.0029; F side x drug (1, 1) = 3.755, p = 0.0705; F knock (1, 1) = 39.51, p < 0.0001; F side (1, 1) = 77.93, p < 0.0001; F drug (1, 1) = 10.02, p = 0.0060) were consistent with the micro-PET/CT finding (Fig. 3B). Furthermore, the results of TH protein were exciting. Western blotting showed the expression of TH protein (three-way ANOVA: F knock x side x drug (1, 1) = 10.06, p = 0.0059; F knock x side (1, 1) = 27.8, p < 0.0001; F knock x drug (1, 1) = 0.3095, p = 0.5857; F side x drug (1, 1) = 9.313, p = 0.0076; F knock (1, 1) = 27.36, p < 0.0001; F side (1, 1) = 23.53, p = 0.0002; F drug (1, 1) = 3.344, p = 0.0862) in the GLT-1-knockdown side of the striatum was reduced compared to their non-AAV-injected side and in the Ctrl group (Fig. 3B), and TH histochemical staining of the striatum also proved this (Fig. 3C). Immunohistochemistry showed that TH-positive neurons in the GLT-1-knockdown side of the SNpc were also reduced compared to their non-AAV-injected side and the Ctrl group (Fig. 3D, three-way ANOVA: F knock x side x drug (1, 1) = 5.161, p = 0.0323; F knock x side (1, 1) = 102.5, p < 0.0001; F knock x drug (1, 1) = 13.81, p = 0.0011; F side x drug (1, 1) = 4.541, p = 0.0435; F knock (1, 1) = 109.1, p < 0.0001; F side (1, 1) = 114.3, p < 0.0001; F drug (1, 1) = 20.17, p = 0.0002). These results suggested that GLT-1 knockdown in unilateral striatal astrocytes could lead to damage to the dopamine system in the substantia nigra-striatum pathway in the knockdown side. Similarly, ceftriaxone blocked all of these changes.
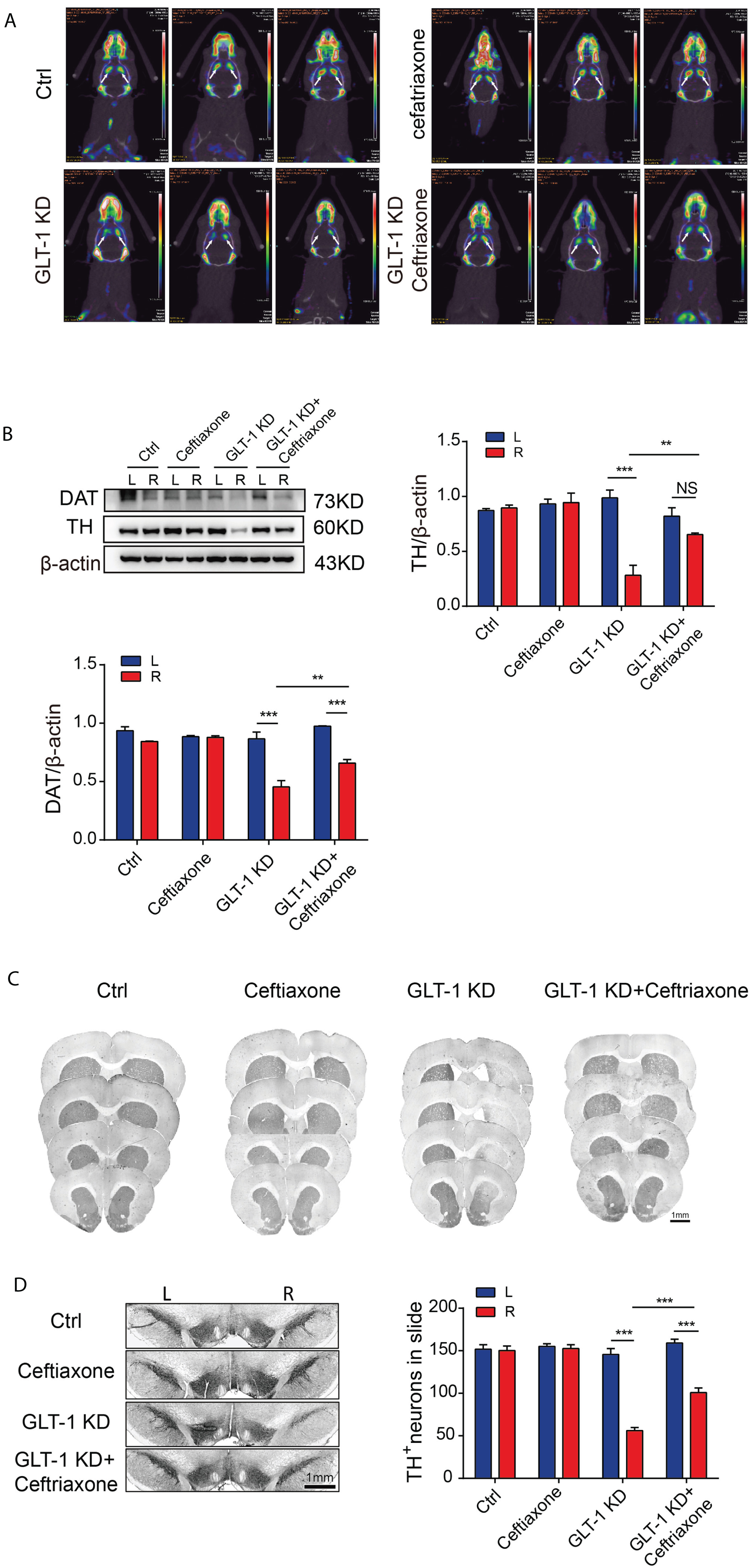
Changes in the dopamine system in the substantia nigra-striatum pathway of SD rats six weeks after knockdown of GLT-1 in right striatal astrocytes in vivo. A) Evaluation of the changes in dopamine transporter in bilateral striatum of rats in the Ctrl, Ceftriaxone, GLT-1 KD and GLT-1 KD + Ceftriaxone groups by micro-PET/CT. B) Expression of DAT and TH protein in bilateral striatum of rats in the Ctrl, Ceftriaxone, GLT-1 KD, and GLT-1 KD + Ceftriaxone groups by western blotting. C) TH histochemical staining of the striatum in rats in the Ctrl, Ceftriaxone, GLT-1 KD, and GLT-1 KD + Ceftriaxone groups (scale bar = 1 mm). D) Immunohistochemistry showed the changes in TH-positive neurons in the SNpc of rats in the Ctrl, Ceftriaxone, GLT-1 KD, and GLT-1 KD + Ceftriaxone groups (scale bar = 1 mm). Bar value = mean±SEM, n = 3. *p < 0.05; **p < 0.01; ***p < 0.001; NS, non-significant.
Knockdown of GLT-1 in primary astrocyte led to morphological damage of TH positive neurons in vitro, which was protected by GLT-1 agonist treatment
We have confirmed using behavioral, histochemical and neuroimaging approaches that in vivo knockdown of GLT-1 in unilateral striatal astrocytes of SD rats can induce Parkinsonian-like changes. In order to further clarify the underlying mechanism and avoid interference by various factors in vivo, we decided to study the effect of astrocytic GLT-1 knockdown on TH-positive mesencephalic neurons in vitro by co-culturing GLT-1-deficient primary astrocytes and normal primary mesencephalic neurons. Firstly, the primary astrocytes and mesencephalic neurons were separated from rats and cultured. We confirmed the identity of primary astrocytes by immunofluorescence staining using the specific marker GFAP and proved that they were almost not mixed with microglia (Fig. 4A). The siRNA sequence targeting GLT-1 was used for transfection of the primary astrocytes, and the knockdown efficacy was evaluated by Western blotting (Fig. 4B, t = 6.838, df = 6, p = 0.0005). Three days the co-culturing GLT-1-knockdown primary astrocytes and primary mesencephalic neurons, we found using immunofluorescent staining and Sholl analysis that the morphology of TH-positive neurons in the si-GLT-1 group was abnormal, mainly manifested as shortened or reduced neurites, while treatment with ceftriaxone protected the neurite of TH-positive neurons (Fig. 4C, Ending radius (two-way ANOVA: F interaction (1, 8) = 2.994, p = 0.1218; F siRNA (1, 8) = 8.624, p = 0.0188; F drug (1, 8) = 45.85, p = 0.0001); Sum inters (two-way ANOVA: F interaction (1, 8) = 4.791, p = 0.0600; F siRNA (1, 8) = 6.003, p = 0.0399; F drug (1, 8) = 23.06, p = 0.0014); Ramification index (two-way ANOVA: F interaction (1, 8) = 0.1497, p = 0.7089; F siRNA (1, 8) = 0.1497, p = 0.7089; F drug (1, 8) = 0.8513, p = 0.3832).
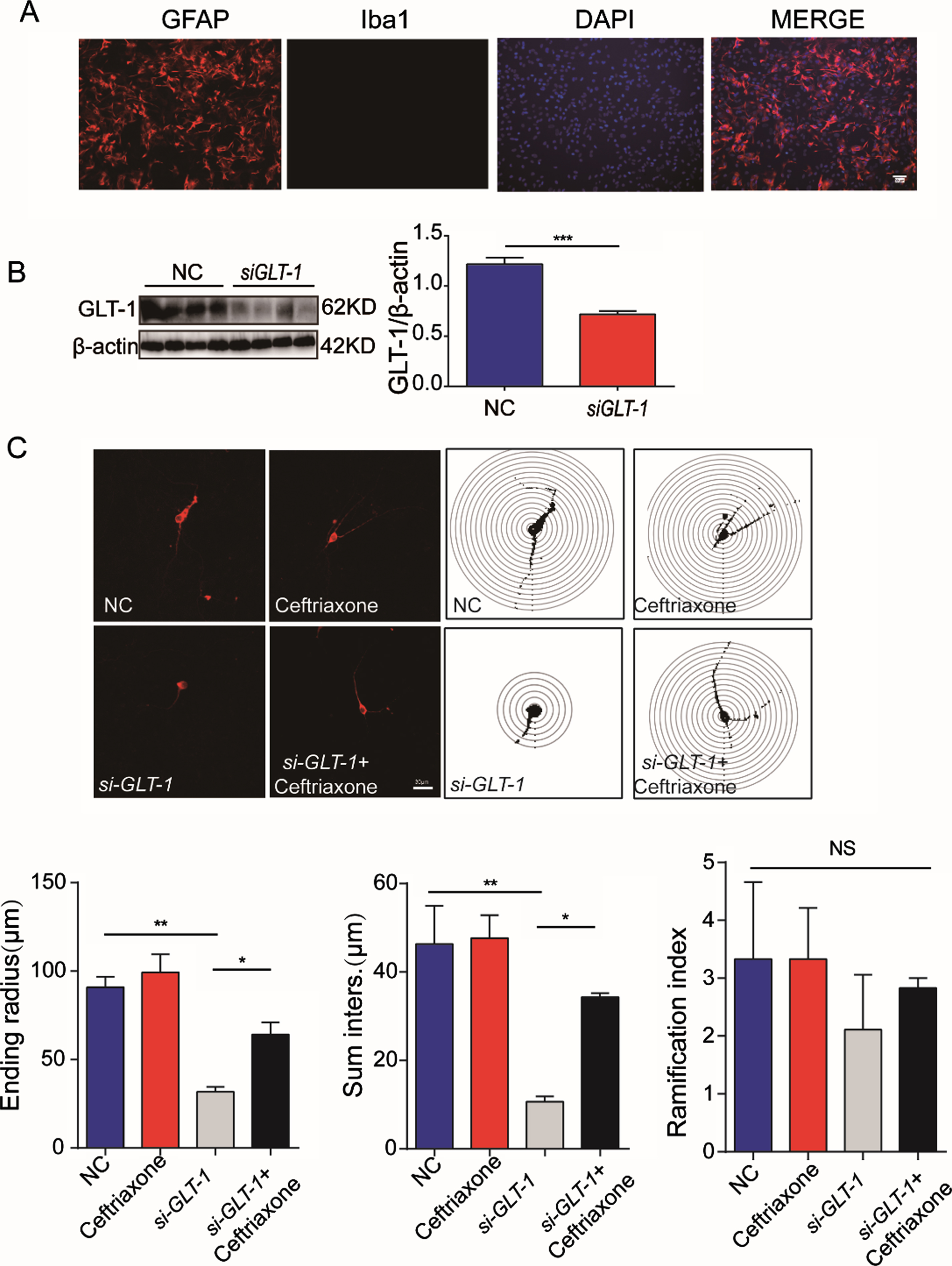
Effect of astrocytic GLT-1 knockdown on morphology of co-cultured TH-positive primary mesencephalic neurons in vitro. A) Immunofluorescence staining of astrocyte marker GFAP, microglia marker IBA1, and nuclear dye DAPI to confirm the identity of primary astrocytes after 14 days in vitro culture and removal of microglia (scale bar = 30μm). B) Western blotting showed the knockdown efficacy of GLT-1-specific siRNA. C) Morphological changes in TH-positive primary mesencephalic neurons co-cultured with primary astrocytes treated with NC, GLT-1 siRNA, or GLT-1 siRNA + Ceftriaxone by immunofluorescence staining and Sholl analysis (scale bar = 30μm). Bar value = mean±SEM, n = 3–4. *p < 0.05; **p < 0.01; ***p < 0.001; NS, non-significant.
Knockdown of astrocytic GLT-1 in vitro provoked accumulation of glutamate in the supernatant and abnormal calcium signaling changes in primary mesencephalic neurons
We demonstrated that TH-positive mesencephalic neurons were damaged by co-culturing with GLT-1-deficient astrocytes in vitro. Which mechanism is involved in this process? Considering the physiological function of GLT-1 and the relevant literatures, we speculated that the following changes might occur after GLT-1 knockdown: GLT-1 deficiency ⟶ dysfunction of glutamate transport in astrocytes ⟶ glutamate accumulation ⟶ neuronal hyperactivity ⟶ abnormal Ca2 + signal in neurons ⟶ neuron injury (Supplementary Figure 2). In order to verify this hypothesis, we first detected the changes in glutamate concentration in the supernatant of the astrocyte-mesencephalic neuron co-culture system using HPLC- pre-column derivatization with OPA, and found increased glutamate in the supernatant after astrocytic GLT-1 knockdown in vitro (Fig. 5A, two-way ANOVA: F interaction (1, 12) = 4.077, p = 0.0664; F siRNA (1, 12) = 6.164, p = 0.0288; F drug (1, 12) = 26.72, p = 0.0002). Furthermore western blotting showed that the expression of p-CaMKII protein in mesencephalic neurons in the astrocytic si-GLT-1 group was higher than that in the NC group (Fig. 5B, two-way ANOVA: F interaction (1, 8) = 9.964, p = 0.0135; F siRNA (1, 8) = 4.007, p = 0.0803; F drug (1, 8) = 27.74, p = 0.0008), suggesting that changes in the regulation of the Ca2 +/CaMK signaling pathway had occurred. As expected, these pathophysiologic disturbances were also restored to a certain extent by the GLT-1 agonist ceftriaxone.
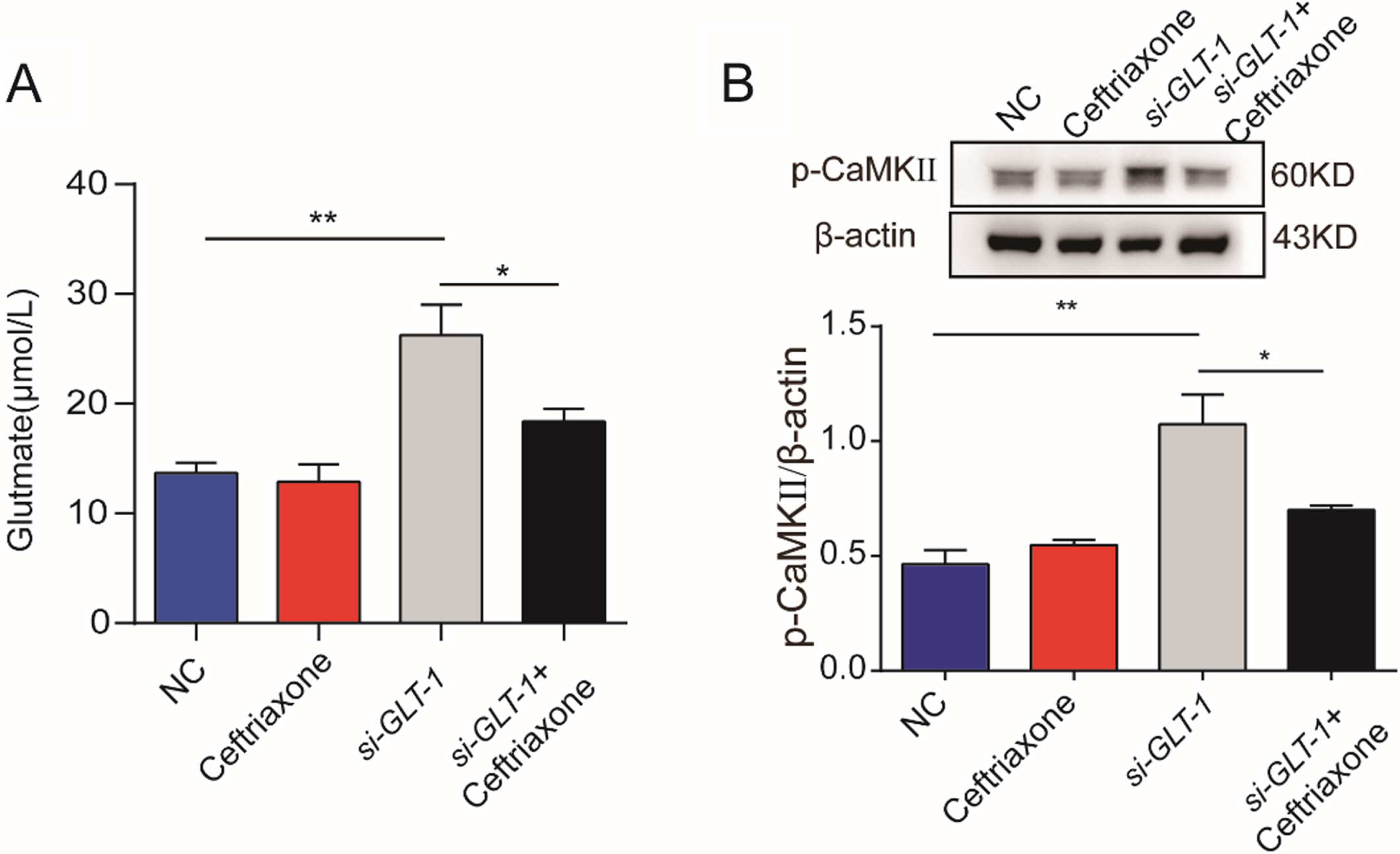
Effects of astrocytic GLT-1 knockdown on glutamate content in the supernatant of astrocyte-mesencephalic neuron co-culture and on intracellular calcium signaling of mesencephalic neurons. A) Glutamate concentration in the supernatant of the co-culture system in the Ctrl, Ceftriaxone, GLT-1 siRNA, and GLT-1 siRNA + Ceftriaxone groups detected by HPLC- pre-column derivatization with OPA. B) Expression of p-CaMKII protein in the Ctrl, Ceftriaxone, GLT-1 siRNA, and GLT-1 siRNA + Ceftriaxone groups detected by western blotting. Bar value = mean±SEM, n = 3–4. *p < 0.05; **p < 0.01.
DISCUSSION
Since the publication of the paper entitled “An Essay on the Shaking Palsy” by the British doctor James Parkinson in 1817 [30], the study of PD has become more in-depth, and reports on the molecular basis of PD are common. Glutamate, an excitatory neurotransmitter, plays a pivotal role in normal basal ganglia circuitry on the one hand and plays an important role in maintaining homeostasis of the SNpc. However, in some pathological conditions, the increased levels of extracellular glutamate and hyperactivation of glutamatergic receptors in the nigrostriatal system trigger a phenomenon called excitotoxicity, which affects cell viability and promote neuronal death. A growing body of evidence has indicated that significant changes in glutamatergic neurotransmission occur during the progression of PD. Alterations in the levels and function of both ionotropic and metabotropic glutamate receptors in the striatum have been described in several different experimental models of PD and in brain tissue from PD patients [31]. In addition, changes in the levels of glutamate transporters were also observed in different animal models of PD [32]. According to the literature, it was determined that the concentration of serum glutamate in PD patients was higher than that in healthy subjects [33]. Thus, altered glutamatergic neurotransmission appears to be central in the pathogenesis of PD. Notably, numerous studies have shown that glutamate-mediated neurotoxicity is not responsible for the initial insult and neuronal loss in the SNpc, but is rather a secondary effect of dopaminergic neurons susceptibility, molecular/bioenergetic defects as well as altered neurotransmission associated with cell death in the nucleus [34]. Previous animal experiments have shown that glutamate receptor related drugs can improve the symptoms of PD; but in the clinic, this is not seen in PD patients due to various side effects. Why do these drugs not prevent and cure PD by targeting glutamatergic neurotransmission? In addition to glutamate receptors, glutamate transporters also plays an irreplaceable essential role in glutamatergic neurotransmission.
As one of the numerous high affinity glutamate transporters (also known as excitatory amino acid transporters, EAATs) encoded by the Slc1a2 gene [35], this transporter called GLT-1 in rodents and EAAT2 in other eukaryotes. Due to its important role in glutamate cycle, discovery of the involvement of GLT-1 in nervous system diseases stemmed from the in-depth study of glutamate excitotoxicity. More and more studies have shown that GLT-1 is related to the development of neuropsychiatric diseases such as depression, schizophrenia, autism, epilepsy, motor neuron diseases (such as amyotrophic lateral sclerosis), synucleinopathies (such as PD), Alzheimer’s disease, Huntington’s disease, multiple sclerosis and chronic pain, and the regulation of GLT-1 may be a novel and effective strategy for the treatment of these diseases [36–40].
Chung et al. [13] first found a reduction in striatal GLT-1 after exposure to 6-OHDA. Zhang et al. [41] found that a decrease in GLT-1 expression caused by abnormal ubiquitination of GLT-1 in an MPTP-induced PD mouse model, which led to motor dysfunction and loss of dopaminergic neurons in mice. Some studies also found that GLT-1 expression decreased in a manganese-induced Parkinsonian-like model [42] and in a DJ-1-mutated PD model [43]. All these finding suggest that the reduction in GLT-1 might be an important risk factor for the occurrence of PD. However, some other studies have found that unilateral injection of 6-OHDA into the medial forebrain bundle could increase the expression of GLT-1 in bilateral striatum in a time-dependent manner [44]. Although it was generally believed that glutamate uptake naturally decreases after GLT-1 downregulation, a study found that the downregulation of GLT-1 by dihydrokainic acid, a specific GLT-1 inhibitor, could lead to increased expression of another glutamate transporter EAAC1 and growing glutamate uptake in the striatum [32]. This contradiction might be explained by the differences in PD models and observation time used, or maybe related to the compensatory mechanism of the body against PD damage. In the disease state, the pathological elevation in glutamate may initially lead to a compensatory increase in GLT-1 expression. However, with progression of the disease, the increase in glutamate overwhelms the compensatory capacity of the body and leads to dysfunction of GLT-1 in glutamate transport, which further aggravates the damage to neurons and astrocytes, leading to the depletion of GLT-1 production by these cells. This eventually causes enhancement of excitatory neurotoxic effects and progression of the disease. Therefore, full clarification of the regulatory mechanism of GLT-1 in PD will help provide a new target for the prevention and treatment of this disease. Unfortunately, up to now, research on the regulatory mechanism of GLT-1 in PD is still very rare. The main reason for this might be the failure in systematic establishment and application of the GLT-1 transgenic PD mouse model. The GLT-1-knockout transgenic mice established in previous studies were either lethal or absent for PD phenotype. In order to solve this problem, some groups have made alternative attempts to achieve suitable models. For example, Assous et al. [45] injected the GLT-1 inhibitor L-trans-pyrrolidine-2,4-ducarboxylate (L-trans-PDC) into rat SNpc, and Zhang et al. [20] injected astrocyte-specific GLT-1-knockdown virus into mouse SNpc, both of which showed a Parkinsonian-like phenotype. However, unfortunately the specific mechanisms involved have not been investigated. In addition, what is more important is that SNpc is not a high-GLT-1 brain area, but the striatum is. Previous studies found that decreased expression of GLT-1 in the PD model often occurs in the striatum, which was also verified in this study. More surprisingly, this decline also occurred in models of PD with depression. Therefore, it is more significant to successfully establish a PD animal model by knocking down the the GLT-1 in striatal astrocytes.
In this study, we stereotactically injected AAV, which specifically transduces astrocytes and knocks down their GLT-1, into two sites in the right striatum of each SD rat. Six weeks after AAV injection, Parkinsonian-like changes were confirmed by behavioral, neuroimaging and histochemical assessments. Not only was motor impairment (poor motor balance, decreased locomotion activity and abnormal gait) observed, but also non-motor features of depression-like behavior. More importantly, we found that there was damage to the dopamine system in the substantia nigra-striatum pathway. These results suggested that our model could mimic some of the motor damage and pathological characteristics of PD similar to previously established models with GLT-1 downregulation in the SNpc [20, 45]. The difference was that our model also showed non-motor symptom of depression-like changes, which suggested that striatal astrocytic GLT-1 might play an important role in PD-associated depression. Although the loss of GLT-1 in astrocytes may account for the alterations in glutamate neurotransmission in depression, so far there is no report of depression caused by direct downregulation of GLT-1 in the striatum. However, it has been reported that reduced GLT-1 was found in the striatum of male stressed-depressed rodents [46], and some scholars have indicated that downregulation and dysfunction of GLT-1 in astrocytes is closely associated with PD-related depression [47]. Collectively, these finding suggest that the role of striatal astrocytic GLT-1 in modulating depression-behavior in PD is worthy of further study. In addition, another difference to be noted was that the time point at which the behavioral change occurred in our model was earlier than that in the model established by Assous et al. [45] and later than that in the model by Zhang et al. [20]. Whether there are other factors leading to this difference is worthy of further exploration in the future. Of course, apart from the above issues, another interesting observation is that in our model the loss of TH-positive neurons in the SNpc was approximately 50%, which was inconsistent with the observation in the previous PD model induced by unilateral injection of 6-OHDA, in which motor injury was triggered only when nearly or over 80%of dopaminergic neurons were lost [48]. One explanation is that, in addition to the difference in the induction substances (neurotoxin 6-OHDA vs. virus), the criteria for behavioral evaluation were also different. Additionally, the effects of non-dopamine system on motor function should also be taken into account. For example, some studies [49, 50] found that dopamine might play a role only in the initiation of movement, but not in the maintenance of the motion. Therefore, the role of glutamate system itself on movement [51], and other neurotransmitter systems than the glutamate-regulated dopamine system, such as the serotonin system, are also worthy of attention. Previous studies have shown that the serotonin system is related to exercise fatigue [52], and GLT-1 may have a regulatory effect on serotonin function [53]. Our own deduction is that our PD model might be in the transition state from the Parkinsonian-like prodromal phase to Parkinsonian-like typical dyskinesia phase. However, it is difficult to draw a definitive conclusion on this issue due to the complex physiological conditions and numerous interfering factors in vivo.
In order to better understand and explain the possible mechanisms involved, we simulated the in vivo condition by in vitro co-culture of primary mesencephalic neurons with primary astrocytes in which GLT-1 was knocked down. Through a morphological study, we confirmed that knockdown of GLT-1 in primary astrocytes caused damage to dopaminergic neurons in vitro, which was consistent with our in vivo findings. Furthermore, histochemically, we found that damage to dopaminergic neurons might be a consequence of the chain reaction of “glutamate accumulation ⟶ neuronal overexcitation ⟶ abnormal intracellular Ca2 + signaling in neurons” following GLT-1 knockdown in astrocytes. We confirmed using HPLC- pre-column derivatization with OPA that increased glutamate content was found in the supernatant of the co-culture after knocking down GLT-1 in astrocytes. According to a previous publication [54], glutamate is normally present in the supernatant of a conventional neuron culture system, but it can be increased by two to three times when neurons are co-cultured with GLT-1 siRNA astrocytes. Our results showed that glutamate was increased approximately two-fold, which is consistent with previous research. Using an immunoblot assay, we further confirmed the increased expression of p-CaMKII protein within the Ca2 +/CaMK signaling pathway in mesencephalic neurons co-cultured with GLT-1-deficient astrocytes. These results on the changes of Ca2+ signaling validated the transcriptome sequencing results of Zhang et al. [20], where they found that the calcium signaling pathway in the SNpc of their Parkinsonian-like mice established by downregulation of GLT-1 in the SNpc was also activated. Thus, our findings once again indicated that the change in calcium signaling might play an important role in the pathological mechanism of PD [55], and the upstream molecule GLT-1 which leads to change in calcium signal, might be an important target for the prevention and treatment of PD.
At present, drugs achieve neuroprotection by regulating GLT-1. Ceftriaxone [56–61], rapamycin [62], iptakalim [63], ginsenoside Rb1 [64], sodium valproate [65, 66], and sodium butyrate [66] have been shown to improve PD-related indicators by upregulating GLT-1 in PD models, among which ceftriaxone and its molecular mechanism is the most extensively studied [60]. In our in vitro study, we also found that the addition of ceftriaxone could prevent morphological damage of dopaminergic neurons induced by GLT-1-knockdown in astrocytes, which was consistent with the in vivo findings that ceftriaxone has a protective effect on the PD rat model induced by GLT-1 knockdown in striatal astrocytes. Unfortunately, due to the restrictions of objective conditions, we were unable to further explore the specific signaling pathway involved in GLT-1 regulation following ceftriaxone treatment in this study.
Although there are a few limitations in this study, we have solved some problems in this field. This study proved that direct downregulation of GLT-1 in astrocytes can lead to Parkinsonian-like changes in vitro and in vivo. We also preliminarily explored the possible mechanism and the protective measures against this change. We hope that our results will shed new light on the future study of the prevention and treatment of PD based on the glutamate excitotoxicity.
Footnotes
ACKNOWLEDGMENTS
This work was supported by Jiangsu Provincial Key R&D Program (BE2018658), the key project of Shandong Provincial Natural Science Foundation (ZR2020KH024), Suzhou Clinical Research Center of Neurological Disease (Szzx2015033), Jiangsu Provincial Medical Key Discipline Project (ZDXKB2016022), Discipline Construction Program of the Second Affiliated Hospital Soochow University (XKTJ-XK202001, XKTJ-RC202004), Suzhou Science and Technology Development Program (SYSD2019114), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.