
Abstract
Background/Objective:
Multiple system atrophy (MSA) is a highly debilitating, rare neurodegenerative disorder with two clinical motor variants (parkinsonian or MSA-P and cerebellar or MSA-C). There is a wide span of motor and non-motor symptoms (NMS) that progress over time. We studied the cohort from the Catalan Multiple System Atrophy Registry (CMSAR) to determine which symptoms are most likely to progress throughout a 2-year follow-up.
Methods:
We analyzed baseline, 12-month, and 24-month follow-up evaluations from the 80 cases recruited by the CMSAR. Evaluations included the UMSARS assessment, cognitive and neuropsychiatric evaluations, and a non-motor scale (NMSS-PD). Statistical analysis was done using a Generalized Estimated Equations (GEE) model.
Results:
Both UMSARS I and II sub-scores significantly increased at 12- and 24-month follow-ups (p < 0.001), with a median total score increase of 11 and 12.5 points, respectively. Items on UMSARS I that significantly worsened were mostly motor affecting daily activities. NMS, including urinary and sexual dysfunction, as well as sleep difficulties showed a significant progression on the NMSS-PD; however, other NMS such as postural hypotension, gastrointestinal, and mood dysfunction, although prevalent, did not show a clear progression on clinical scales.
Conclusion:
Within 24 months and as early as 12 months, MSA cases may experience significant motor worsening, affecting basic daily activities. NMS are prevalent; however, not all clinical scales register a clear progression of symptoms, perhaps suggesting that they are not sensitive enough for non-motor evaluation.
INTRODUCTION
Multiple system atrophy (MSA) is a synucleino-pathy, with two main motor phenotypes, either pres-enting as parkinsonism (MSA-P) or as cerebellar ataxia (MSA-C). Motor symptoms do not respond well to levodopa, and most patients become wheel-chair-bound a few years after diagnosis [1, 2], with a mean survival rate of 8-9 years. Both phenotypes share a wide range of non-motor symptoms (NMS), particularly, but not solely, significant autonomic dysfunction, which is essential for a possible/probable diagnosis according to consensus guidelines [3]. Alt-hough postural hypotension (OH) is the most characteristic sign, urinary symptoms followed by sexual dysfunction are the most frequent [4].
The Unified Multiple System Atrophy Rating Sc-ale (UMSARS) was designed to appropriately examine all MSA cases reliably and consistently and has been validated for use in clinical trials and studies [5]. There are four sections which include a clinical history inquiring about daily activities, as well as autonomic and motor symptoms (UMSARS I–sco-ring from 0–48), a physical examination (UMSARS II- scoring from 0–56), blood pressure measurements (UMSARS III), and an overall disability score (UMSARS-GDS-scoring from 1–5). The two largest prospective studies to date, one North American (NA) [6] and the other European (EMSA) [4]) used this scale to assess over 300 total cases combined, and both reported a clear progression of the disease represented as a significant annual mean UMSARS score increase of over 30%. However, these studies did not specifically describe which items were driving the changes, which is the most relevant clinical outcome for a patient. Other NMS were not studied either, as the UMSARS only collects information from autonomic symptoms, but symptoms such as mood, cognition, or sleep are not represented.
We led the multicenter Catalan Multiple system atrophy registry (CMSAR), intending to recruit and assess a significant number of MSA cases in a sta-ndardized manner. A complete clinical characterization of the disease is important to understand how the disease progresses to help predict clinical outcomes for patients and clinical-trial assessments. Thus, this paper aims to describe how a patient diagnosed with MSA progresses over two years and evaluates which scales best reflect these changes.
METHODS
We recruited MSA patients from March 2015-March 2018, referred from different movement dis-orders units from Catalonia, and included them in the CMSAR. This program is a multicenter observational and prospective clinical registry of MSA patients, approved by the ethics committee at Hospital Clinic of Barcelona, including clinical scales accompanied by a bio-repository of bio-samples collected through standardized protocols [7]. Subjects provided written informed consent and were included if they met the second consensus clinical criteria for possible or probable MSA-P or MSA-C subtypes [3]. This was supported by clinical examination, medical records, and diagnostic tests provided by their usual neurologist. All baseline and follow-up evaluations were conducted by a neurologist who coordinated the CMSAR and were supervised by two movement disorders experts with broad experience in atypical parkinsonisms (MJM) and cerebellar ataxias (EM). Standardized data was collected and recorded through a centralized case report form data capture. Personal information was recoded to preserve confidentiality. A structured neurological examination included the UMSARS assessment (I, II, III, and GDS) [5], as well as the non-motor scale in Parkinson’s disease (NMSS-PD) [8], a quality of life scale (EuroQol-5D) [9], the Beck Depression Inventory (BDI-II) [10], Frontal Assessment Battery (FAB) [11], Mini-Mental State Examination (MMSE) [12], Mattis Dementia-Rating Scale (MDRS-2) [13], REM Sleep Behavior Disorder Screening Questionnaire (RBD-SQ) [14], and the Epworth Sleepiness Scale (ESS) [15]. For the UMSARS III section, blood pressure measurements were taken in supine, followed by standing position at three-minute intervals. If cases were physically unable to maintain these positions for at least 3 minutes, the measurement was not recorded. A brief description of all scales is included in Supplementary Table 1.
Statistical analysis
Results are described using median and 95%confidence interval (CI) for quantitative variables and absolute frequencies and percentages for qualitative variables. Since this is a longitudinal observati-onal study, Generalized Estimated Equations (GEE) models were made to account for intrasubject correlation employing an autoregressive matrix assumption (AR-type 1). This model handles longitudinal data providing a correct estimation of the parameters with a more precise calculation of standard errors and correct inferences [16]. All analyses using GEE models were applied after rank transformation of dependent variables to obtain a non-parametrical approach. In all statistical tests, we applied a two-sided type I error of 5%. Statistical calculations were made using SPSS version 25. Since this is a general approach to evaluate MSA progression, all statistical analyses have been presented without adjustments for multiplicity; thus, p-values are interpreted as nominal for descriptive purposes. Due to the study’s prospective nature, the baseline data includes excluded cases by reason of intention to treat (ITT) concept. By doing so, our results reflect a real-life scenario and are clinically applicable.
RESULTS
General demographics
We recruited 80 cases with possible (n = 40) or probable (n = 40) MSA (53%male, 54%MSA-P variants) (Table 1). A total of 52 cases completed a one-year follow-up (12 m-FU), and 42 completed a two-year follow-up (24 m-FU) (Fig. 1). Loss of follow-up was mainly due to voluntary withdrawal from the study (16 subjects) and death (15 subjects). Six of these cases underwent brain donation, confirming a definite diagnosis of MSA. During follow-up reassessments, seven cases were excluded owing to reasonable diagnostic doubts. The main differential diagnoses included Parkinson’s disease (PD) and other atypical Parkinsonisms. A reanalysis of the data excluding these cases, did not show any relevant changes.
Demographic and clinical scale data at baseline for the whole cohort and by clinical variants at 12 (12 m-FU) and 24-month follow-up (24 m-FU): Data shown as median scores for all clinical scales and confidence intervals at 95%. *indicates a significant change (p < 0.05) as evaluated by GEE (adjusted by disease duration), at one year (Δ1y) or two years (Δ1y) follow-up, or a significant difference between MSA-P variants and MSA-C variants (PvsC)
y, years; M, male; F, female; m, months; nc, non computable; LEDD, levodopa equivalent daily dose; UMSARS, Unified MSA rating scale; GDS, Global disability scale; NMSS-PD, non-motor scale in Parkinson’s disease; EuroQoL-5D, European quality of life scale 5 dimensions; BDI, Beck Depression Inventory; MMSE, Mini-Mental State Examination; MDRS-2, Mattis Dementia-Rating Scale; FAB, Frontal Assessment Battery; RBD-SQ, REM-sleep behavior disorder screening questionnaire; ESS, Epworth sleepiness scale.
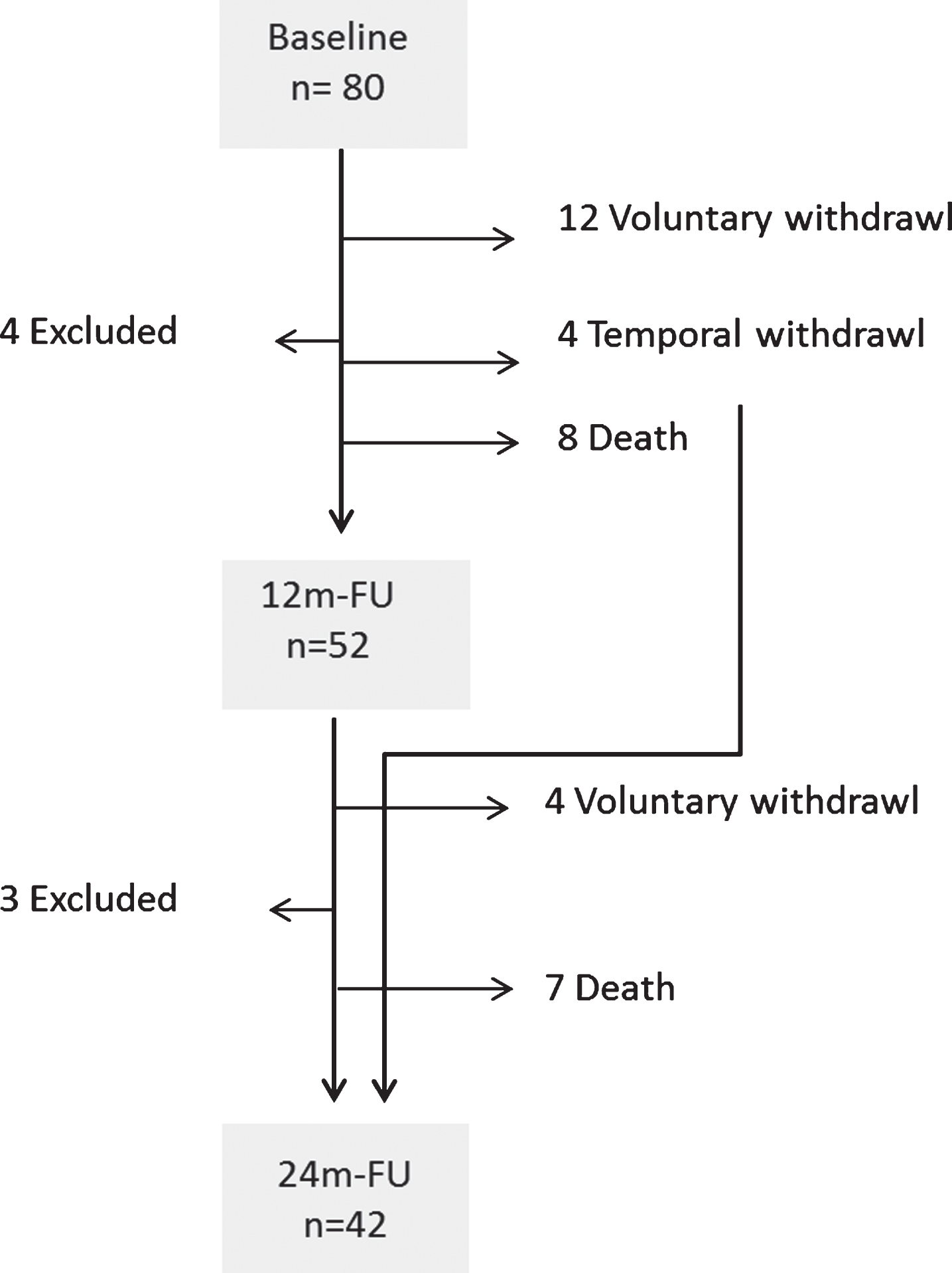
Diagram representing total cases recruited and causes for loss of follow-up: Number of subjects (n) at each follow-up interval, including baseline visit, 12 months follow up visit (12 m-FU), and 24-month follow-up visit (24 m-FU). Causes for loss of follow-up include voluntary withdrawal, temporal withdrawal (unable to attend temporarily but did complete the subsequent visit), death, and exclusion by a neurologist (Excluded) due to diagnostic doubts.
GEE models were made for the crude effect of any type of MSA and by MSA-P or MSA-C as fi-rst diagnosis, as well as the effect adjusted by disease duration and by age of onset (appearance of first motor symptom). The age of onset did not significantly change results; thus, data seen on tables corresponds to results adjusted by disease duration, showing an effect. MSA-P and MSA-C cases did not show any significant differences regarding age, gender, age at onset, or disease duration; however, GDS scores were higher in MSA-P cases.
UMSARS evaluation
Both UMSARS I and II sub-scores significantly increased at 12 m-FU and 24 m-FU (p < 0.001) with a median total (I and II) score increase of 11 and 12.5 points respectively (23%and 27%change). When analyzing the progression of UMSARS’ total scores in cases with a mild disease stage (GDS score ≤2) at baseline (n = 44), there was a median total score increase of 14 points at 12 m-FU and 22 points at 24 m-FU (41%and 64%change). On the other hand, moderate to severe cases (GDS ≥3) had a median total UMSARS score increase of 11 points at 12 m-FU and 17 points at 24 m-FU; however, there were only 12 cases from the original 36 at 24 m-FU.
Items that significantly worsened on the UMSARS I were mostly motor. The only non-motor item that significantly increased at both intervals was urinary dysfunction (p = 0.018 and p < 0.001) (Fig. 2). Items on UMSARS II that worsened at 12 m-FU and 24 m-FU were related to limb movements, speech, and postural reflexes. Gait and rigidity significantly worsened after 24 m-FU. MSA-P cases showed a significantly higher UMSARS II score when compared to MSA-C cases (p = 0.004) with higher scores at baseline for items related to rest tremor (p = 0.007), rigidity (p < 0.001), alternate movements (p < 0.001), finger (p < 0.001) and heel tapping (p < 0.001) as well as posture (p < 0.001). On UMSARS I, MSA-P cases also showed higher scores at baseline for items related to difficulties with eating (p = 0.046), dressing (p = 0.013), and intestinal dysfunction (p = 0.023). A subsequent analysis evaluating clinical variants adjusted by GDS scores showed that when assessing mild MSA-P vs. mild MSA-C cases, there were no significant differences in eating and dressing difficulties, and the total UMSARS score was no longer significantly higher in MSA-P cases. MSA-C cases only scored higher baseline scores on UMSARS II item 3 (oculomotor findings, p = 0.027). The p-values for the longitudinal changes of each variant and item can be seen in Table 2.
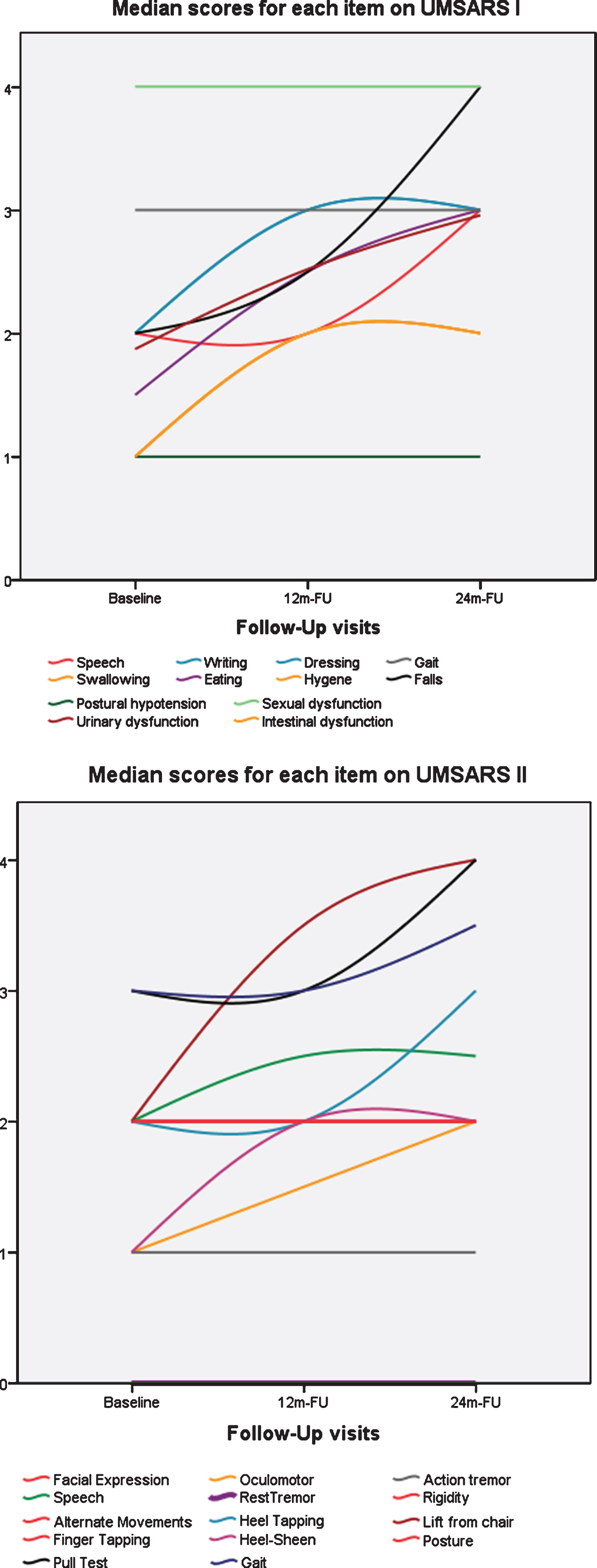
Progression of items on UMSARS assessment. Median Scores for each item on UMSARS I (above) and UMSARS II (below) at baseline, one-year follow-up (12 m-FU), and two-year follow-up (24 m-FU). Note that most non-motor symptom-related items on UMSARS I do not show clear progression.
UMSARS evaluations with P-values indicating significant change for each item. Arrows indicate the direction of change (↑ indicates score increase, ↓ indicates decrease, Ø indicates no significant change) for each item score between either 12 or 24month follow-up scores (12 m-FU or 24 m-FU) compared to baseline visit along with the corresponding p-value. As evaluated by GEE, a significant change for disease duration was considered when the p-value was < 0.05
UMSARS, Unified MSA rating scale; OH, postural hypotension; GU, genitourinary system; dys, dysfunction; Sex, sexual; GI, gastrointestinal; AT, action tremor; Alt movs, alternate movements; FT, finger tapping; HT, heel tapping; HS, heel-sheen.
A total of 72 cases underwent UMSARS III at baseline, 45 at 12 m-FU, and 33 at 24 m-FU. Severe orthostatic hypotension (SOH) (SOH≥30mmHG or DOH≥15) was recorded 27 times in 21 subjects (26%of the cohort). Only 13 of these subjects had either SOH or DOH at baseline. SOH or DOH appeared at 12 m-FU in six subjects and two more subjects at 24 m-FU.
UMSARS-GDS (global disability scale) significantly worsened at 12 m-FU and 24 m-FU (p < 0.001) (Table 1).
NMSS-PD evaluation
NMS, as assessed by the NMSS-PD, mainly in-cluded sexual, urinary, and gastrointestinal dysfunction. However, consistent and significant changes were only seen in the sleep, urinary, and sexual dysfunction subsections. MSA-P cases scored higher than MSA-C cases in the sleep (p = 0.02, higher reports for diurnal somnolence and fatigue), gastrointestinal (p = 0.018, higher scores for dysphagia and drooling), and miscellanea (p = 0.019, scored higher on weight change) subsections (Table 3). Diplopia became significantly more frequent in MSA-C at 12 m-FU than MSA-P (p = 0.013) cases.
NMSS-PD median score data by subsections of the whole cohort and by clinical variant. Data represents the median scores with 95%confidence intervals for each subsection of the NMSS-PD assessment at 3-time points (baseline, 12 months or 12 m-FU, and 24 months or 24 m-FU). *indicates a significant change (p < 0.05) as evaluated by GEE adjusted by disease duration, at 12 m-FU (Δ1y) or 24 m-FU (Δ2y) follow-up or a significant difference between MSA-P variants and MSA-C variants (PvsC).
Other evaluations
There were no significant differences at 12 m-FU or 24 m-FU when assessing BDI, MMSE, MDRS-2, or FAB scales. Median scores for MDRS-2 assessment, however, suggest that most cases have mild cognitive difficulties. Regarding sleep scales, the median RBD-SQ scores suggest that the majority of cases have REM-sleep behavior disorder. This scale showed a significant total score decrease at 24 m-FU (p = 0.01). The ESS median score does not suggest significant diurnal somnolence (score <10), though there was a significant increase at 24 m-FU (p = 0.009). Nocturnal stridor was reported in 21 subjects (26%) at baseline, which increased to 46%(n = 37) after two years. MSA-P cases scored higher than MSA-C cases at baseline on GDS (p = 0.023) and ESS (p = 0.003). EuroQoL-5D scores suggest a low quality of life for most cases with a median score of 13, which was significantly higher for MSA-P than MSA-C cases at 12 m-FU (p = 0.028) (Table 1).
At baseline visit, 25 cases (31%) required a whe-elchair for safe transportation. At 12 m-FU and 24 m-FU, 14 and 12 new cases were wheelchair users. Thus, 64%of the original cohort required a wheelchair after a two-year follow-up. A urinary catheter was required at some time point during the two years in 22 cases (27.5%). Three cases required a percut-aneous endoscopic gastrostomy (PEG). Clinical phenotypes were equally distributed among all intervals for both wheelchair dependency and urinary catheter use; however, 73%of cases using a urinary catheter were males.
Medication
The levodopa equivalent daily dose (LEDD) was higher in MSA-P cases at all intervals (p < 0.002); however, the dose did not significantly change thr-oughout two years. Other common medications taken during the study included antidepressants (57.5%of the cohort) and benzodiazepines (mostly clonaz-epam), taken by 60%of the cohort. Treatment for postural hypotension (mostly fludrocortisone) was taken by 41%of cases, and agents for urinary symptoms (due to overactive bladder) were taken by 17.5%.
DISCUSSION
MSA is a highly debilitating and complex disease with a broad spectrum of motor features and NMS. When diagnosed, many patients ask about what they should expect, and once symptoms begin to progress, many questions arise regarding quality of life and prognosis. During our two-year follow-up study, we indeed report a clear progression of motor symptoms and NMS reflected on the UMSARS I, II, and IV (GDS), as well as on the NMSS-PD. The UMSARS assessment was the most sensitive scale, and we found that significant motor changes were appropriately recorded. However, although prevalent, the progression of NMS was not well reflected on any scale. Work is warranted to register the progression of these symptoms in MSA appropriately.
When considering the rate of change on the UMSARS assessments, our results are similar to previous studies, with a slightly lower rate of change. By assessing each item independently, we found that the increase in points is mostly related to the motor items on the UMSARS assessments. Though NMSS-PD scores significantly increased, there were few consistent changes regarding NMS. Other scales used to assess neuropsychiatric symptoms, such as BDI and cognitive assessments (MMSE, FAB, and MDRS-2), did not significantly increase throughout the 12 m-FU and 24 m-FU either. It is important to point out that when studying the median scores, we see that symptoms such as depression are present at baseline (median score on BDI of 16.5) and continue to be so throughout two years (median score of 17.5), despite antidepressant treatment. Curiously, typical autonomic symptoms such as postural hypotension, sexual dysfunction, and constipation were present in both UMSARS and NMSS-PD assessments; however, these symptoms did not necessarily show pr-ogression on these scales. This may be because symptoms appear early in the disease and plateau quite rapidly or that these scales are not sensitive eno-ugh to evaluate further changes or worsening (ceiling effect) (Fig. 2). Subjective memory complaints such as forgetfulness or loss of concentration significantly increased at 24 m-FU, and MDRS-2 scores suggested mild cognitive difficulties, especially in the initiation section (testing verbal fluency). Progression of these symptoms was not reflected on cognitive scales such as MMSE, FAB, and MDRS-2, suggesting that these scales do not detect the specific and mild cognitive changes that these cases may undergo in 2 years.
Urinary symptoms progressed on both UMSARS and NMSS-PD assessments. These symptoms may be a differential factor between other similar diseases, such as PD. An overactive bladder is common to both pathologies with joint symptoms, including urinary urgency, nocturia, and increased urinary frequency. However, the severity of these symptoms is usually the main difference between diseases, with faster progression to urinary incontinence in MSA cases [17]. In fact, at baseline, 37.5%of our cases had urinary incontinence, which increased to 54%at 12 m-FU, and 64%at 24 m-FU. This is clearly in contrast to the presence of severe OH. Severe OH is a specific clinical sign for this disease, albeit not as frequent as urinary dysfunction. For example, although most cases had some degree of OH (median SOH of –10 mmHg), a significant drop was only found in 26%of the cohort. These low values may be in relation to medication use (taken by 30%of the cohort) and the increased variability of this finding. Also, significant OH is probably underrepresented when assessing under UMSARS guidelines. Perhaps prolonged monitoring of blood pressure would have been more sensitive, as seen in the EMSA study [4], where 54%of cases had moderate (≥20 mmHg) or severe OH within 3 min, which increased to 72%within 10 min. This states the importance of extending the monitoring of blood pressure for a more accurate diagnosis.
Regarding sleep changes, over 60%of cases scored over 5 points on the RBD-SQ, which is suggestive of REM-sleep behavior disorder (RBD) at baseline and 12 m-FU. The total score was significantly lower after 24 m-FU (though 48%of cases still scored over 5). The meaning of this is uncertain as this questionnaire was created as a screening test rather than a progression scale. Also, we should consider that most cases (around 60%) are treated for their RBD, and as the disease progresses, many bed partners no longer share beds, which may underrate results. Of note, at baseline, 31 subjects had undergone a video-polysomnography (VPSG) to evaluate their sleep disorder, and 27 were suggestive of RBD (2 of these cases scored under 6 on the RBD-SQ). VPSG results also reported stridor in 7 cases (3 had not been reported during the clinical interview). This suggests that VPSG remains the gold standard.
Concerning phenotypic differences, both clinical variants in our cohort worsened at 12 m-FU and 24 m-FU, and over 60%of cases required a wheelchair after two years. MSA-C cases experienced a higher increase of falls compared to MSA-P cases at 12 m-FU and 24 m-FU, and also had more oculomotor findings at baseline, 12 m-FU, and 24 m-FU, as well as diplopia affecting daily leisure activities such as reading and watching TV. These oculomotor differences are in keeping with other studies (17). Our EuroQoL-5D results, however, suggested that quality of life may be affected earlier in MSA-P cases, which also scored higher on the UMSARS II evaluation and had more difficulties with daily activities on UMSARS I. In a retrospective study by Watanabe et al. [2] with 230 Japanese cases, MSA-P patients also showed a more rapid functional deterioration than MSA-C patients, with similar survival. In the European trial, shorter survival was found for MSA-P variants; however, no differences in survival were found in the North American (NA) trial. Thus, though some reports have postulated a worse prognosis for MSA-P variants [4, 18], many others have not [6, 19–22]. This data suggests that MSA-P cases may be more functionally affected than MSA-C cases, though this does not necessarily translate into a shorter survival.
Our results indicate that the UMSARS assessment is probably the most valuable clinical tool to assess the progression of motor symptoms. Other scales such as BDI, MMSE, NPI, FAB, and MDRS may not be appropriate when studying NMS in a cohort of these dimensions. Nevertheless, the UMSARS assessment holds several caveats, especially concerning a possible ceiling effect in many items. When looking at Fig. 2, one can see that only a few items show clear progression (mainly items related to postural instability). Gait, for example, does not seem to progress when examined by clinical history in contrast to when assessed by physical examination. As for NMS, only urinary dysfunction clearly progresses, whereas there is no progression regarding postural hypotension or sexual dysfunction. Moreover, we must keep in mind that the UMSARS assessment was created to assess both variants with the same scale; and in this sense, we find it appropriately evaluates the overall severity among both variants since GDS scores correlate with UMSARS and quality of life scores. However, specific clinical differences may be overlooked, possibly being more sensitive to parkinsonian symptoms. Perhaps a simplified and less biased assessment is warranted to track progression more accurately. We must also take into account that the rate of change depends on disease duration and disease stage. Cases with GDS scores of 1 or 2 at baseline showed a higher rate of change than those enrolled with a GDS score of at least 3. This again implies that cases may reach a clinical plateau after two years or that the UMSARS assessment has a ceiling effect after this time.
The previous European and North-American prospective efforts [4, 6] provided important information on MSA progression, mainly focusing on the rate of change captured by the UMSARS and survival. Nevertheless, to the best of our knowledge, this is the first prospective MSA cohort study describing how motor and NMS progress using several clinical scales in an integrated manner. Study limitations include a small sample size at 24-month follow-up, which is common to all MSA prospective studies since MSA is a rare disease, and as the disease progresses, a considerable amount of subjects pass away or become too disabled to attend follow-up visits. Consequently, this may create a possible selection bias as cases that complete 24 m-FU may be more benign; however, by using a GEE model, this bias is reduced significantly since the data given is an estimation taken from the complete cohort baseline data matrix. Another selection bias is that many cases were advanced (45%) with a median disease duration of 4 years. As seen in our results, progression rates may be different in earlier-stage patients. A cohort of only early-stage patients may show different results. Clinical criteria should be updated to try to improve diagnosis at these earlier stages. In addition, not all scales have been validated for MSA patients; however, part of our study was to assess if these were valuable for prospective studies. Also, other valuable scales such as COMPASS-31 [23] or SCOPA-AUT [24] have not been included, as well as more specific scales to study the cerebellar components of MSA-C such as ICARS [25]. Finally, diagnostic certainty is always an explicit limitation in MSA; yet the repeated follow-up visits with continuous evaluation by movement disorder experts help ensure that most cases will probably have this disease, and 6/6 cases were confirmed by autopsy. Of note, 7 cases were excluded from the registry due to considerable diagnostic doubts, especially with PD and other atypical Parkinsonisms. Results do not relevantly change when excluding these cases; however, we chose to analyze by ITT concept to predict how a patient with an MSA diagnosis will progress after two years, even if misdiagnosed. Autopsy studies indicate that about 20%[26] to 40%[27] of MSA cases are misdiagnosed. This clearly demonstrates the urgent need for more diagnostic tools to confirm the diagnosis in vivo, especially at early stages.
CONCLUSIONS
MSA patients should expect a significant motor worsening in two years, generally affecting all daily activities and postural stability. The UMSARS assessment is a sensitive scale to study motor progression at one and two-year follow-up visits but may have a ceiling effect. NMS are also prevalent and mostly affect sexual, urinary, and gastrointestinal functions as well as sleep and mood; however, NMS require a more specific and sensitive scale to study progression. This broad spectrum of difficulties affects quality of life and remains mainly untreated.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
Footnotes
ACKNOWLEDGMENTS
We would like to thank all the patients for their always willing and generous collaboration. All this has been possible thanks to the funding from the Fundació Marató TV3 and CERCA Programme from Generalitat de Catalunya.