
Abstract
Progressive supranuclear palsy (PSP) is an atypical parkinsonism with prominent 4R-tau neuropathology, and the classical clinical phenotype is characterized by vertical supranuclear gaze palsy, unprovoked falls, akinetic-rigid syndrome and cognitive decline. Though PSP is generally regarded as sporadic, there is increasing evidence suggesting that a series of common and rare genetic variants impact on sporadic and familial forms of PSP. To date, more than 10 genes have been reported to show a potential association with PSP. Among these genes, the microtubule-associated protein tau (MAPT) is the risk locus with the strongest effect size on sporadic PSP in the case-control genome-wide association studies (GWAS). Additionally, MAPT mutations are the most common cause of familial PSP while the leucine-rich repeat kinase 2 (LRRK2) is a rare monogenic cause of PSP, and several other gene mutations may mimic the PSP phenotype, like the dynactin subunit 1 (DCTN1). In total, 15 MAPT mutations have been identified in cases with PSP, and the mean age at onset is much earlier than in cases carrying LRRK2 or DCTN1 mutations. GWAS have further identified several risk loci of PSP, proposing molecular pathways related to PSP. The present review focused on genetic studies on PSP and summarized genetic factors of PSP, which may help to elucidate the underlying pathogenesis and provide new perspectives for therapeutic strategies.
Keywords
INTRODUCTION
Progressive supranuclear palsy (PSP), a rare neurodegenerative disorder with a prevalence of approximately 5–7 per 100000 [1], is traditionally considered to be one of the most common atypical parkinsonian syndromes and increasingly recognized to involve a range of motor, behaviour and language abnormalities [2]. PSP is clinically heterogeneous and presents as different phenotypes [3, 4], among which the most classic phenotype is Richardson’s syndrome (PSP-RS, also known as Steele–Richardson–Olszewski syndrome), which was first described as a clinicopathological entity in 1964 [5, 6]. New clinical diagnostic criteria of PSP (MDS-PSP) were published in 2017 [7], improving the sensitivity of PSP, in particular the variant PSP clinical presentations [8], and proposing four degrees of diagnostic certainty, namely, definite PSP, probable PSP, possible PSP and suggestive of PSP [7].
The pathological features of PSP are a predominance of 4-repeat (4R) tau inclusions in the form of neurofibrillary tangles, neuropil threads, tufted astrocytes and oligodendroglial coiled bodies in basal ganglia, diencephalon and brainstem, mainly affecting the globus pallidus, subthalamic nucleus and substantia nigra, in addition to neuronal loss and gliosis [9, 10]. PSP is the most common primary 4R-tauopathy, and the neuropathological 4R-tau begins to abnormally accumulate during the presymptomatic phase [2]. The localization of tau pathology is a major drive of clinical heterogeneity [4], and the distribution and severity of tau pathology vary in different clinical phenotypes, suggesting the significance of further studies into the pathological processes related to PSP [11, 12].
Although PSP is generally recognized as a sporadic syndrome, there are familial forms of PSP [13–15] and few pedigrees with PSP-like phenotypes [16, 17], and a pattern of autosomal dominant inheritance has been proposed [14, 15]. A case-control study has observed more first-degree relatives with parkinsonism or dementia in patients with PSP than in controls, demonstrating familial aggregation in PSP [18]. In contrast, genome-wide association studies (GWAS) in large cohorts over the past few years have identified several loci significantly associated with PSP [19–22], prompting studies on PSP genetics. Though a series of questions remain to be solved, genetics play an important role in PSP. This review focused on genetic studies and summarized genetic factors associated with PSP, especially recent additions, aiming to better understand PSP at the genetic level.
SINGLE-GENE MUTATIONS ASSOCIATED WITH PSP
MAPT in PSP
The microtubule-associated protein tau (MAPT) gene, encoding the tau protein, is located on chromosome 17q21.31 and consists of 16 exons [23]. Among the exons, exons E0 and 14 are non-coding, and exons 4a, 6 and 8 are not transcribed or expressed in human brain [24]. Alternative splicing of exon 10 produces two major isoforms of tau protein, namely 4R-tau and 3R-tau, with four and three microtubule binding repeats, respectively. In adult human brain, the ratio of 4R to 3R is roughly equal to 1 [25]. Zero, one or two N-terminal inserts result from alternative splicing of exon 2 or exon 2 and 3 together, and each N-terminal insert contains 29 amino acids. Therefore, there are six major tau isoforms with different lengths in the human brain (Fig. 1) [24]. MAPT mutations were first reported in families of frontotemporal dementia (FTD) and parkinsonism linked to chromosome 17 (FTDP-17) in 1998 [26]. Since then, more than 60 mutations in MAPT have been identified, mostly characterized by behavioural changes and/or clinical parkinsonism [27]. MAPT is involved in a series of neurodegenerative disorders [28] and the case-control GWAS of PSP has identified that MAPT is the risk locus with the strongest effect size [19].
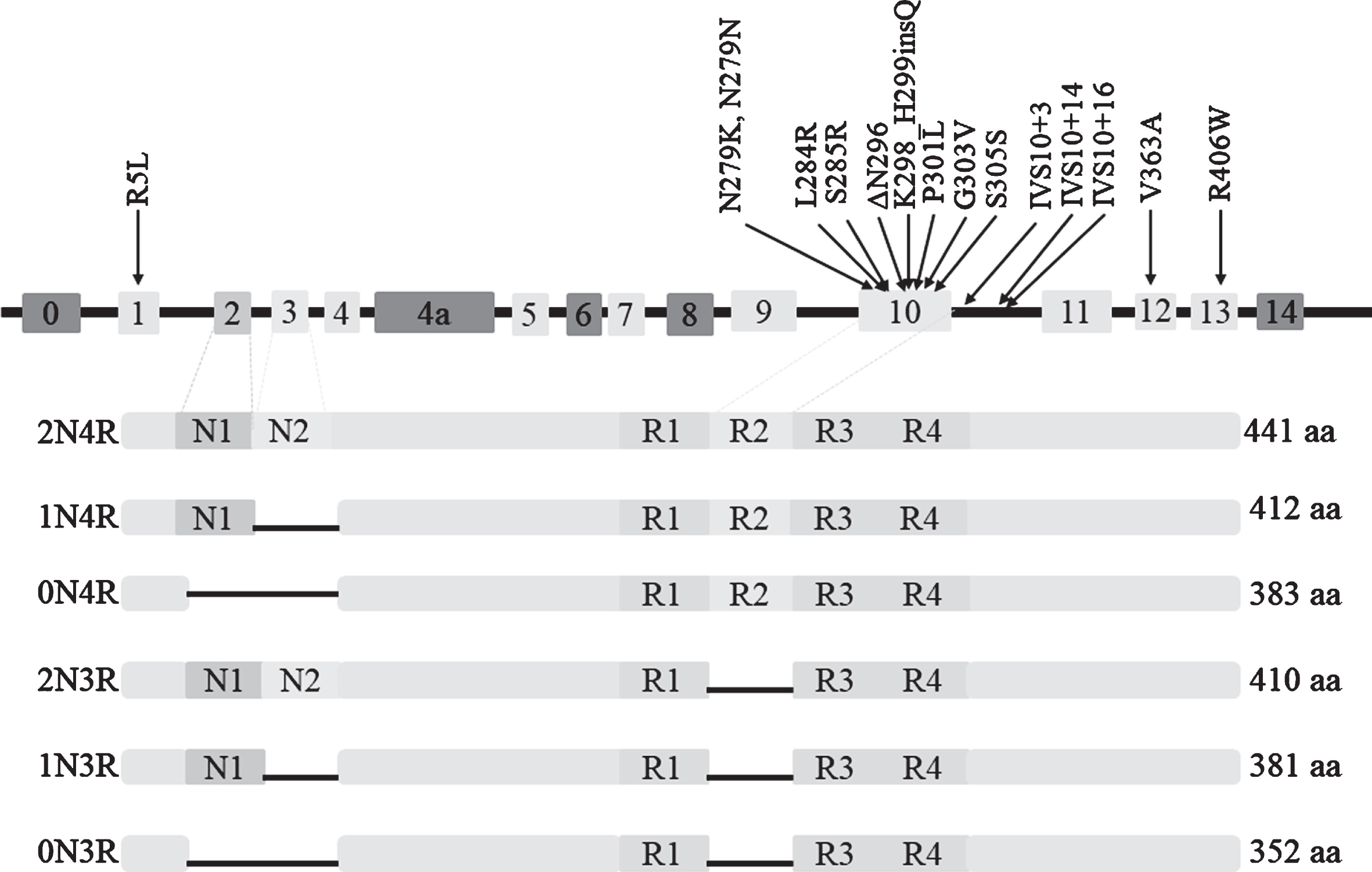
Six isoforms of protein tau and 15 mutations of MAPT associated with PSP.
MAPT haplotypes and PSP
A case-control study first observed that the homozygous A0 alleles containing 11 TG repeats in intron 9 of MAPT are overrepresented in PSP, identifying a dinucleotide repeat polymorphism linked with PSP and providing direct genetic evidence for the association between PSP and MAPT [29], which has been validated by several groups [30–32]. Baker et al. [33] subsequently found that the polymorphic dinucleotide marker is inherited in complete disequilibrium linkage with other eight common single nucleotide polymorphisms (SNPs). These researchers initially described two extended haplotypes (called H1 and H2) covering the entire MAPT and observed that haplotype H1 with 238 bp in intron 9 is overrepresented in PSP patients compared to controls. Various studies on Caucasian populations have emphasized the significant association between MAPT haplotype H1 and PSP [34, 35]. A GWAS directly compared rs8070723 to H1/H2 as a proxy and observed that the odds ratio (OR) for PSP in H1/H1 carriers in the GWAS is 5.46 (p = 1.5×10–116) [19], and the GWAS suggested that the H2 haplotype had a protective role [36].
The structure of the H1 haplotype has been explored, and over 20 common subhaplotypes have been identified. Among which, MAPT H1c is significantly overrepresented in patients with PSP and in patients with corticobasal degeneration (CBD) [36] and Alzheimer’s disease (AD) [37]. H1c increases MAPT expression, especially 4R-containing transcripts [38], but underlying mechanisms remain unclear. Using a large series of autopsy-confirmed PSP cases, Heckman et al. identified three other MAPT H1 subhaplotypes associated with PSP, namely H1g, H1d and H1o, and they also proposed that several subhaplotypes may play roles in the severity of tau pathology in PSP [39], further advancing the understanding of the H1 haplotype.
MAPT mutations and PSP
The frequency of PSP cases carrying MAPT mutations is various with a range from 0.6% to 14.3% [16, 41]. In total, 15 different mutations of MAPT have been described in cases presenting as PSP (clinical or neuropathological diagnosis), as shown in Table 1 and Fig. 1. Except for the R5L mutation in exon1, the V363A mutation in exon12, and the R406W mutation in exon13, other causative mutations exist in exon 10 and its splicing regions, resulting in an increased ratio of 4R/3R. The most common MAPT mutation causing PSP is located at codon 279, involving 11 cases. The p.K298_H299insQ in exon 10 has been identified in three patients with familial PSP through genetic sequencing of 165 cases with possible tauopathies, and this mutation is the first insertion mutation reported in MAPT. Yasuda et al. identified a N279K mutation in a Japanese patient with pallido-nigro-luysian degeneration (PNLD) [42], which is pathologically distinctive from typical PSP and is considered a variant of PSP [43].
MAPT mutations causing PSP-like syndrome
No., number; AAO, age at onset; NA, not available; PSP, progressive supranuclear palsy; PNLD, pallido-nigro-luysian degeneration; FTDP-17, frontotemporal dementia and parkinsonism linked to chromosome 17; AD, Alzheimer’s disease; PSP-AD, PSP with concomitant AD. “PSP-like” pathology means that tau accumulations exist in brain but do not meet the pathological diagnostic criteria of PSP. The letter “f” in clinical diagnosis indicates the case with a family history of parkinsonism, dementia, or other neurodegenerative diseases.
The onset of PSP is insidious, presenting with different symptoms, which highlights the clinical heterogeneity and complexity of PSP as well as the necessity of follow-up. The mean age at onset (AAO) in PSP with MAPT mutations is approximately 44.8 years (range of 36–62 years) with a peak in the early 40s. Two families [44, 45] carrying the ΔN296 mutation had an earlier age (<40 years) when first PSP-related symptoms occurred compared to cases carrying other mutations. Moreover, Pastor et al. [45] identified a patient carrying the homozygous ΔN296 mutation, which is the first case carrying a pathogenic homozygous mutation in MAPT, resulting in a more severe phenotype compared to heterozygous mutations, the latter causing a milder condition with reduced penetrance.
Except for the R5L mutation described in a sporadic PSP case, Table 1 shows that the majority of cases with other MAPT mutations have a family history with parkinsonism, dementia, or other neurodegenerative disorders, further supporting the familial aggregation in PSP, which is consistent with the results reported by Kaat et al. [18]. This phenomenon indicates the importance to exclude MAPT mutations when such a family history exists.
Recently, Chen et al. observed 2 patients with autopsy-confirmed PSP harboring duplications spanning the entire MAPT locus (both copy number = 3) through genome-wide survey of copy number variants (CNVs) and proposed that MAPT duplications may be a genetic cause of PSP, which provides genetic evidence for the hypothesis that CNVs are associated with PSP [22] and indicates that there are more potential associations between MAPT and PSP.
LRRK2: a very rare monogenic cause of PSP pathology
Leucine-rich repeat kinase 2 (LRRK2) is considered as one of the most common genetic causes of Parkinson’s disease (PD), and genetic evidence has shown more than 5 LRRK2 mutations linked with both familial and sporadic PD [59–61]. Although several studies have detected negative results when screening LRRK2 in PSP cohorts [62–65], 5 mutations have been identified in patients presenting a PSP phenotype (Table 2). A recent study identified that common variation at the LRRK2 locus is a genetic determinant of PSP survival (Jabbari et al. unpublished data), adding further evidence for the association between LRRK2 and PSP.
LRRK2 mutations in cases with PSP phenotype
PSP, progressive supranuclear palsy; AD, Alzheimer’s disease; AAO, age at onset; NA, not available; “+”, present; “–”, absent.
The R1441C mutation has been reported in a large kindred named family D with pleomorphic pathology and within 10 affected members who are clinically characterized by parkinsonism, but only one member has pathological changes similar to PSP. Thus, the researchers speculated that LRRK2 might contribute to tauopathy in addition to synucleinopathy [66]. The R1441H mutation has been identified in a patient originally diagnosed as typical PD but transiting to PSP 8 years later. This unusual case indicates that the R1441H is involved in the PSP-Parkinsonism (PSP-P) [67], but the involvement of environmental factors in this process remains unknown. G2019S has been observed in 4 cases with PSP from different studies [59, 68–70], and functional in vitro studies have observed that the G2019S mutation increases kinase activity, potentially explaining how LRRK2 causes neurodegeneration [71]. Trinh et al. [72] observed the T2310M mutation in a patient with PSP when sequencing LRRK2, and they also identified 27 other rare nonsynonymous variants in the cohorts. A novel p.A1413T mutation has been identified in a case-control study containing 1039 PSP and 145 CBD patients, and this mutation has been predicted to be “disease damaging” by several in silico predictive algorithms [59].
Therefore, we conclude that the LRRK2 gene is a rare pathologic gene associated with PSP [59] despite inconsistent results in some studies. More association studies in larger cohorts and various populations are needed to further elucidate how these LRRK2 mutations lead to tauopathy and whether other mutations increase the risk for PSP.
Single-gene mutations in cases mimicking PSP
DCTN1 mutations in cases as PSP look-alike syndromes
Dynactin subunit 1 (DCTN1), which encodes p150glued, is the largest subunit of dynactin complex, and it is involved in microtubule binding and molecular transport [74]. DCTN1 mutations have been identified in families with motor neuron disease [75] and Perry syndrome [76], which is a rare autosomal dominant neurodegenerative disease. Unlike tauopathy in PSP, the underlying pathology in DCTN1 mutation carriers is transactive response DNA-binding protein of 43 kDa (TDP-43) proteinopathy [77]. To advance the understanding of clinical phenotype spectrum related to DCTN1 mutations, we summarized 3 DCTN1 mutations reported in patients with a PSP look-alike syndrome (Table 3). However, the p.R1261Q mutation in one PSP-PS case and the p.L896V mutation in one PSP-FTD case both detected by Yabe et al. [16] are not included in Table 3 due to lack of clinical data.
DCTN1 mutations associated with PSP look-alike syndromes
PSP, progressive supranuclear palsy; AAO, age at onset; NA, not available; “+”, present.
Table 3 shows that the mean AAO among these cases is 56.3 years with a large range of disease duration. All patients presented parkinsonism as a major clinical feature, and symmetrical frontal atrophy was obvious in more than half of the cases. DCTN1 mutations play a role in susceptibility to PSP. Due to the low frequency and limited knowledge, however, the underlying mechanism between DCTN1 and tauopathy remains unclear and needs further validation.
Other genes and PSP look-alike syndromes
As genetic studies on neurodegenerative disorders develop, more genes have shown potential links with PSP. A new series of gene mutations or polymorphisms have been identified in patients with a PSP look-alike phenotype, including but not limited to the NPC1 gene [81, 82], the C9orf72 gene [40, 83], the parkin gene (PARK2) [84–86], the transactivation response element DNA-binding protein gene (TARDBP) [87, 88], the progranulin gene (GRN) [16, 89], the TANK-binding kinase 1 gene (TBK1) [90], and the bassoon gene (BSN) [16] (Table 4). Some of these genes have been considered as causative genes in certain neurodegenerative diseases [91–94]. Considering the wide phenotype spectrum of monogenic mutations and the overlap among clinical features of neurodegenerative diseases [95], there may be common mechanisms and pathways in the pathogenesis of parkinsonian syndromes.
Single-genes associated with PSP look-alike syndromes
*The case carried a compound heterozygous mutation (P1007A and F1221fsX20).
In summary, cases carrying MAPT mutations have an earlier AAO (mean: 44.8 years), and the initial features are variable, mainly presenting parkinsonism, unstable walking and frontal cognitive/behavioural symptoms. In addition, a positive family history exists in most cases. The AAOs in patients with LRRK2 mutations are much older (mean: 72.3 years), and the main clinical presentation is parkinsonism with a baseline clinical diagnosis of PD in some cases. Similarly, the typical symptoms in cases carrying DCTN1 mutations are parkinsonism (mean AAO: 56.3 years) and brain atrophy. The relationships between genes and phenotypes may help clinicians to correctly diagnose. Although the associations between genes and PSP require more validations and investigations, further genetic screening is necessary when MAPT mutations are absent, especially in familial cases reminiscent of PSP.
COMMON VARIANTS IDENTIFIED THROUGH GENOME-WIDE APPROACHES
Melquist et al. carried out a pooled genome-wide scan of 500288 SNPs in 2007 and identified chromosome 11p12-p11 as another major risk locus for PSP following the MAPT haplotype H1. They further narrowed the locus to rs901746, the top ranked SNP, which mainly encompasses two genes (DNA damage-binding protein 2 (DDB2) and lysosomal acid phosphatase 2 (ACP2)) [97].
GWAS have identified thousands of genes and SNPs that contribute to complex diseases in humans [98] since the first GWAS was published in 2005 [99], introducing a new perspective on genetic studies and showing promising values in clinical applications [100]. the first large PSP GWAS revealed that MAPT rs8070723 and rs242557 are strongly associated with PSP (1.5×10–116 and 4.2×10–70, respectively), and it uncovered 3 novel risk loci of PSP (shown in Table 4) [19], highlighting new directions and strategies for PSP studies. Subsequent GWAS consisting of European cohorts identified additional variants (Table 5).
Risk loci identified through GWAS
Chr, chromosome; SNP, single nucleotide polymorphism; OR, odds ratio; CI, confidence interval; NA, not available; PSP, progressive supranuclear palsy; Ref., reference; NC, normal controls; GWAS, genome-wide association study; STX6, Syntaxin 6; EIF2AK3, Eukaryotic translation initiation factor 2 alpha kinase 3; MOBP, Myelin-associated oligodendrocyte basic protein; DUSP10, Dual specificity phosphatase 10; SLCO1A2, Solute carrier organic anion transporter family member 1A2; TRIM11, tripartite motif-containing protein 11; RUNX2, runt-related transcription factor 2.
GWAS have not only identified genetic factors likely increasing the risk for PSP but have also identified several molecular pathways contributing to PSP pathogenesis [19, 101]. Eukaryotic translation initiation factor 2 alpha kinase 3 (EIF2AK3) encodes the pancreatic endoplasmic reticulum kinase (PERK) protein, which is involved in the endoplasmic reticulum unfolded protein response (UPR) [102]. Activated UPR is present in regions affected in PSP, and UPR activation is related to tau [103]. Furthermore, loss of PERK function due to EIF2AK3 mutations causes neurodegeneration-like changes, including tau pathology [104]. Because tau normally does not traffic through the endoplasmic reticulum, the mechanisms involved in PERK, UPR and tauopathies are unknown [19]. Surprisingly, the increased representation of rs1768208 with the minor T-allele in PSP cases is more closely linked with the expression of the SLC25A38/Appoptosin gene, though its location is nearer to the myelin-associated oligodendrocyte basic protein (MOBP) gene [19, 105]. Zhao and his collaborators showed that appoptosin overexpression contributes to tau cleavage via caspase activation, which results in aggregation of insoluble tau, disruption of synaptic structures, and deficits of motor function in tau transgenic mice [105], confirming the associations among rs1768208, appoptosin and PSP as well as suggesting a potential diagnostic biomarker for tauopathies.
A GENETIC DETERMINANT OF PSP PHENOTYPE
Jabbari and colleagues carried out a clinical phenotype GWAS through comparing PSP-RS cases to non-RS cases in PSP cohorts, and suggested that rs564309, an intronic variant of the tripartite motif-containing protein 11 gene (TRIM11), may be a genetic modifier of clinical phenotype in PSP [106]. This is the first clinical phenotype GWAS in PSP and opens up new directions for the roles that genetics play in PSP phenotypes. On the other hand, as previous study has proposed that TRIM11 has a critical role in the clearance of misfolded proteins via ubiquitin proteasome system (UPS) [107], Jabbari’s study further proves that UPS is involved in tau pathology, which may provide a target for PSP therapy [106].
CONCLUSION
As a complex disorder involving multiple factors, PSP is challenging due to the inexplicit pathogenesis, lack of effective medications and poor prognosis. The genetic background of PSP has gained growing attention, and many data have suggested that genetics play a role in the susceptibility to PSP and that mutations of certain genes (such as MAPT) directly lead to PSP pathology. Faced with a series of risk factors and biological pathways associated with PSP, larger cohorts are required to validate these associations in addition to identify more novel loci. Functional studies are urgently needed to further elucidate underlying mechanisms, thus introducing new perspectives for diagnostic biomarkers and therapeutic targets for PSP and other tauopathies.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
Footnotes
ACKNOWLEDGMENTS
This study was supported by the National Natural Science Foundation of China (No. 81671075, No. 81971029 and No. 81701134), the National Key R&D Program of China (No. 2017YFC0840100 and 2017YFC0840104), the Provincial Key Plan for Research and Development of Hunan (No. 2017SK2031).