
Abstract
Background:
We recently showed that striatal overexpression of brain derived neurotrophic factor (BDNF) by adeno-associated viral (AAV) vector exacerbated L-DOPA-induced dyskinesia (LID) in 6-OHDA-lesioned rats. An extensive sprouting of striatal serotonergic terminals accompanied this effect, accounting for the increased susceptibility to LID.
Objective:
We set to investigate whether the BDNF effect was restricted to LID, or extended to dyskinesia induced by direct D1 receptor agonists.
Methods:
Unilaterally 6-OHDA-lesioned rats received a striatal injection of an AAV vector to induce BDNF or GFP overexpression. Eight weeks later, animals received daily treatments with a low dose of SKF82958 (0.02 mg/kg s.c.) and development of dyskinesia was evaluated. At the end of the experiment, D1 and D3 receptors expression levels and D1 receptor-dependent signaling pathways were measured in the striatum.
Results:
BDNF overexpression induced significant worsening of dyskinesia induced by SKF82958 compared to the GFP group and increased the expression of D3 receptor at striatal level, even in absence of pharmacological treatment; by contrast, D1 receptor levels were not affected. In BDNF-overexpressing striata, SKF82958 administration resulted in increased levels of D1–D3 receptors co-immunoprecipitation and increased phosphorylation levels of Thr34 DARPP-32 and ERK1/2.
Conclusion:
Here we provide evidence for a functional link between BDNF, D3 receptors and D1–D3 receptor close interaction in the augmented susceptibility to dyskinesia in 6-OHDA-lesioned rats. We suggest that D1–D3 receptors interaction may be instrumental in driving the molecular alterations underlying the appearance of dyskinesia; its disruption may be a therapeutic strategy for treating dyskinesia in PD patients.
INTRODUCTION
Dyskinesia is a major complication of long-term L-DOPA treatment that develops in the vast majority of parkinsonian patients. Previous studies have suggested that brain derived neurotrophic factor (BDNF) plays a role in the appearance of dyskinesia, at least in animal models of Parkinson’s disease (PD) [1, 2]. Accordingly, we have recently shown that striatal overexpression of BDNF, obtained by an adeno-associated viral (AAV) vector, exacerbated L-DOPA-induced dyskinesia (LID) in unilateral 6-OHDA-lesioned rats. This effect was accompanied by a sustained sprouting of striatal serotonergic terminals, which may account for the worsening of LID [1]. Indeed, serotonergic neurons are known to contribute to the appearance and maintenance of LID by providing a site of L-DOPA conversion to dopamine and unregulated neurotransmitter release in L-DOPA-treated hemiparkinsonian rats [3, 4]. Thus, by inducing sprouting of striatal serotonergic terminals, BDNF overexpression has been suggested to worsen LID through an exacerbation in synaptic dopamine swings [1].
The role of BDNF in dyskinesia may not be limited to the trophic effect exerted on the serotonergic system; in fact, BDNF has been reported to affect a number of molecular events that could potentially alter dopamine receptor signaling, and therefore, the development of dyskinesia. Interestingly, Guillin and colleagues reported that BDNF from corticostriatal neurons induces behavioral sensitization to L-DOPA by triggering overexpression of dopamine D3 receptors (D3R) in the striatum of hemiparkinsonian rats [5, 6]. Similar results were obtained in MPTP-treated monkeys, where LIDs were accompanied by striatal overexpression of D3R and administration of a D3R partial agonist strongly attenuated dyskinesias [6].
In light of these reports, the present study investigated whether BDNF overexpression was able to affect striatal expression of D3R and aggravate the development of dyskinesia. In order to circumvent the role of serotonergic terminals in mediating L-DOPA-derived dopamine receptor activation, we used the selective dopamine D1 receptor (D1R) agonist SKF82958 instead of L-DOPA. Postmortem investigations aimed at clarifying the molecular mechanisms accounting for the behavioral effects.
MATERIALS AND METHODS
Animals
Adult male Sprague–Dawley rats (275–300 g; Envigo, Huntingdon, UK) were used in the present study. Rats were group-housed (4–5 animals per cage) (bedding Lignocel ® 3/4S, Envigo, Huntingdon, UK) in an environment maintained at a constant temperature and humidity with a 12 h light/dark cycle (07 : 00 a.m. lights on) and free access to food (4RF21, Mucedola, Settimo Milanese, Italy) and water. All rats were handled daily by experimenters and all experiments followed the ARRIVE guidelines of Laboratory Animal Care [7]. The procedures used were in accordance with the European and Italian legislation on the use and care of laboratory animals (EU Directive 2010/63 and Italian D.Lgs. 2014/26). Animal care and experimental protocols were approved by the Italian Ministry of Health. All efforts were made to minimize the number of animals used and their suffering.
Drugs
All drugs were diluted in 0.9% sterile saline. 6-OHDA and SKF82958 were purchased from Sigma-Aldrich. The dose of SKF82958 was chosen based on a pilot study in order to induce only few dyskinetic movements in the GFP control 6-OHDA-lesioned rats.
Production of the recombinant AAV viral vector
Transfer plasmids carrying adeno-associated viral (AAV)5 inverted terminal repeat coding for either rat BDNF or enhanced green fluorescent protein (GFP) downstream of a cytomegalovirus enhancer hybrid synthetic chicken β-actin (CBA) promoter were generated. Transfection into HEK 293 cells was carried out using the calcium phosphate method and included the appropriate transfer plasmid encoding-enhanced BDNF or GFP and the packaging plasmids pDP5 encoding for the AAV5 capsid proteins. Cells were transfected with 2.5 μg of DNA with equimolar amounts of helper and transfer DNA. Transfected cells were incubated for 3 days before being harvested in phosphate buffered saline–EDTA. The cell pellet was treated with a lysis buffer (50 mM Tris, 150 mM NaCl, pH 8.4) and lysed through freeze–thaw cycles in a dry ice/ethanol bath. The lysate was then treated with 21 U/ml benzonase (Sigma) for nuclear digestion. The crude lysates were purified first by ultracentrifugation (1.5 h at 350,000 g at 18°C) in a discontinuous iodixanol gradient and the virus-containing fractions were purified with ion-exchange chromatography using fast protein liquid chromatography. The virus suspension was then concentrated using a concentrator (Millipore Amicon Ultra, 100 kDa molecular weight cut-off) at 1500×g and 18°C in two consecutive steps by adding phosphate buffered saline. The physical titres of recombinant AAV5 vectors were determined using dot blot quantification as described previously [8]. Genome copy titres were determined using real-time quantitative polymerase chain reaction PCR, and the following vector concentrations were used: 1.2×1014 and 3.1×1013 genome copies/ml for BDNF and GFP, respectively. The AAV-GFP vector was injected as a control at a dilution that matched the number of genome copy per ml of the transgene of interest.
Surgical procedures
Experimental parkinsonism
All 6-OHDA injections were conducted using a stereotaxic frame with an attached Hamilton syringe and under general anesthesia, induced by i.p. injection of a 20 : 1 mixture of Fentanest (Pfitzer, Italy) and Domitor® (Orion Pharma, Italy) at a volume range of 1.4–1.6 ml. Animals received 6-OHDA injection into the right medial forebrain bundle (16 μg free base in 4 μl in 0.02% L-ascorbic acid in 0.9% saline), at the following coordinates relative to the bregma [9]: AP: –4.4 mm, ML: –1.2 mm; DV: –7.8 mm from the dura surface, in order to achieve a complete lesion of the ipsilateral nigrostriatal pathway. Injection speed was 1.0 μl/min and the syringe was kept in place for an additional 3 min before it was slowly retracted.
AAV vector injection
Surgical procedure for AAV vector injection was performed under general anesthesia, as used for 6-OHDA injections. In order to reduce the damage to the brain and donor cells, a glass capillary (outer diameter 60–80 μm) was fitted onto the needle of a 5 μl Hamilton syringe. Rats received 4 μl (2 μl/site; 1 μl/deposit) of the AAV-BDNF or AAV-GFP solution into the 6-OHDA-lesioned striatum at the following coordinates relative to the bregma [9]: AP/ML = +1/–2.8 mm, DV = –5/–4 mm; AP/ML = +0.2/–4 mm, DV = –5.5/–4.5 mm from the dura surface. Injection speed was 0.5 μl/min and the needle was left in place for an additional 3 min period before it was slowly retracted [1].
Experimental design
First, animals were lesioned with 6-OHDA injected into the medial forebrain bundle (n = 60). Three weeks later, rats were subjected to the stepping test in order to estimate their forepaw impairment; only rats with a severe deficit (stepping test score for the parkinsonian paw between 0 and 4 for forehand plus backhand directions) were allocated into the study (n = 54; Supplementary Figure 1A) [10]. One week later, animals were divided into two groups and subjected to the striatal injection of an AAV vector overexpressing either BDNF or GFP, ipsilaterally to the lesion. The dilutions of the vectors were chosen based on our previous study (i.e., 3% for BDNF, 5% for GFP) [1]. After eight weeks, treatment with SKF82958 (0.02 mg/kg/day s.c.) began. A cohort of rats (n = 18) injected with the BDNF or GFP AAV vector was treated for 8 days with SKF82958 (0.02 mg/kg/day s.c.; AAV–GFP + SKF and AAV–BDNF + SKF, n = 9/group) and then rats were perfused for immunohistochemical analyses of tyrosine hydroxylase (TH), BDNF, or GFP expression under general anesthesia, as described for 6-OHDA injections (Fig. 1A). In order to study the possible neurochemical modifications in dopaminergic signaling induced by repeated low dose SKF82958 administration and, at same time, to avoid the possible development of adaptive phenomena, a second cohort of rats, injected with BDNF (n = 18) or GFP AAV vector (n = 18), was treated for 4 days with saline or SKF82958 (0.02 mg/kg/day) and sacrificed by decapitation 20 min after the last injection. The right striata were collected and stored at –80°C for biochemical studies (AAV–GFP + Saline; AAV–GFP + SKF, AAV–BDNF + Saline, AAV–BDNF + SKF, n = 9/group; Fig. 2). This timing was based on a preliminary experiment showing development of clear-cut dyskinetic movements at this time.
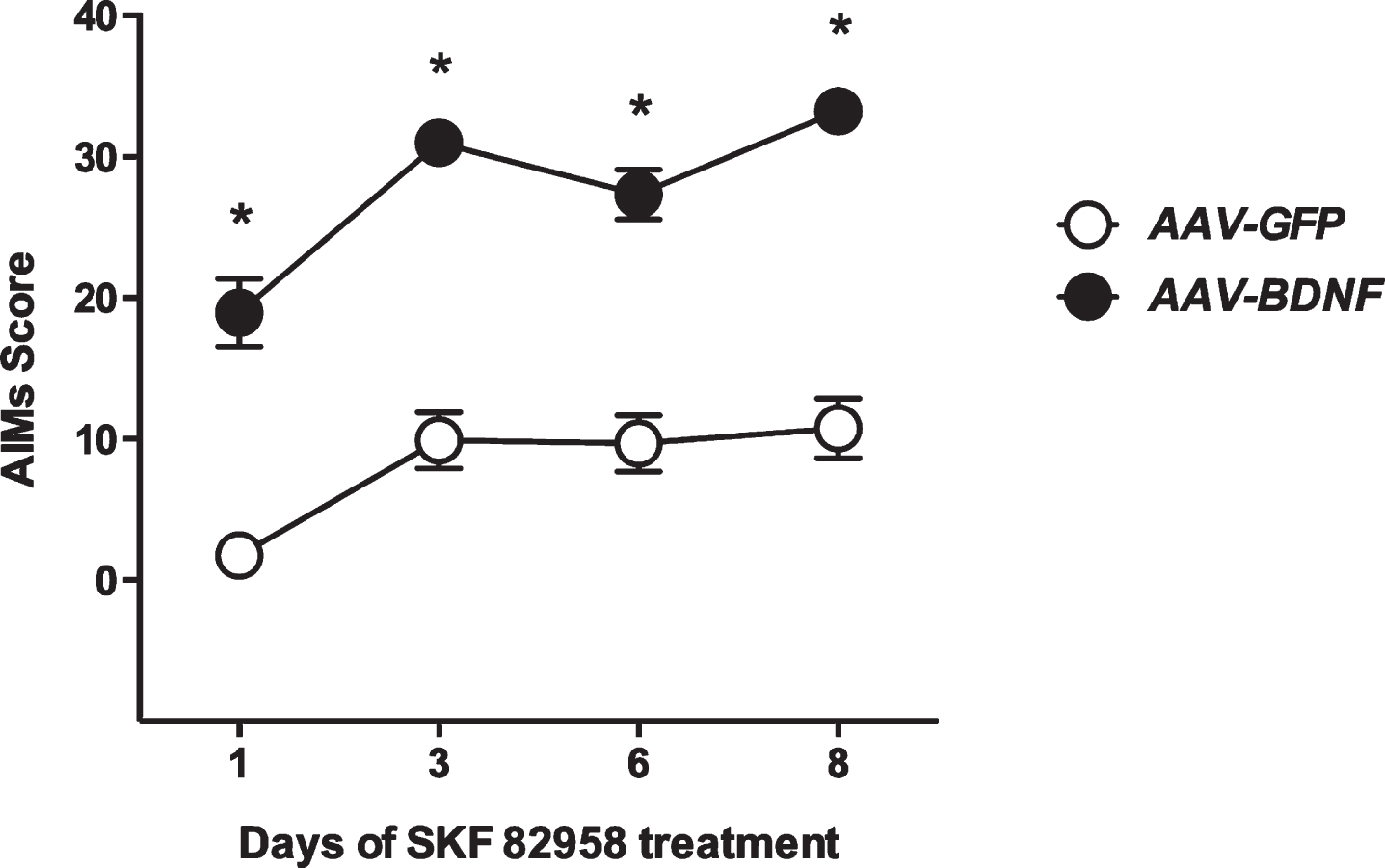
Effect of the D1 receptor agonist SKF82958 (0.02 mg/kg/day s.c.) administration on AIMs score in 6-OHDA lesioned rats injected with either striatal AAV–BDNF or AAV–GFP vectors. Animals were first lesioned with 6-OHDA injected into the medial forebrain bundle and three weeks later they were subjected to the stepping test in order to estimate their forepaw impairment. One week later, animals were divided into two groups and injected with an AAV vector overexpressing either BDNF or GFP in the striatum, ipsilaterally to the lesion. After eight weeks, AAV–GFP (n = 9) and AAV–BDNF (n = 9) injected rats received SKF82958 (0.02 mg/kg/day s.c.) for 8 days, and were tested for the development of abnormal involuntary movements (AIMs). Scores are shown as the sum of orolingual, limb, and axial abnormal involuntary movements (AIMs) in 120 min. Values are expressed as mean±SEM for each experimental group (n = 9/group). Two-way ANOVA for repeated measures followed by Bonferroni’s post hoc test: *p < 0.0001 vs the same time point of the AAV–GFP + SKF group.
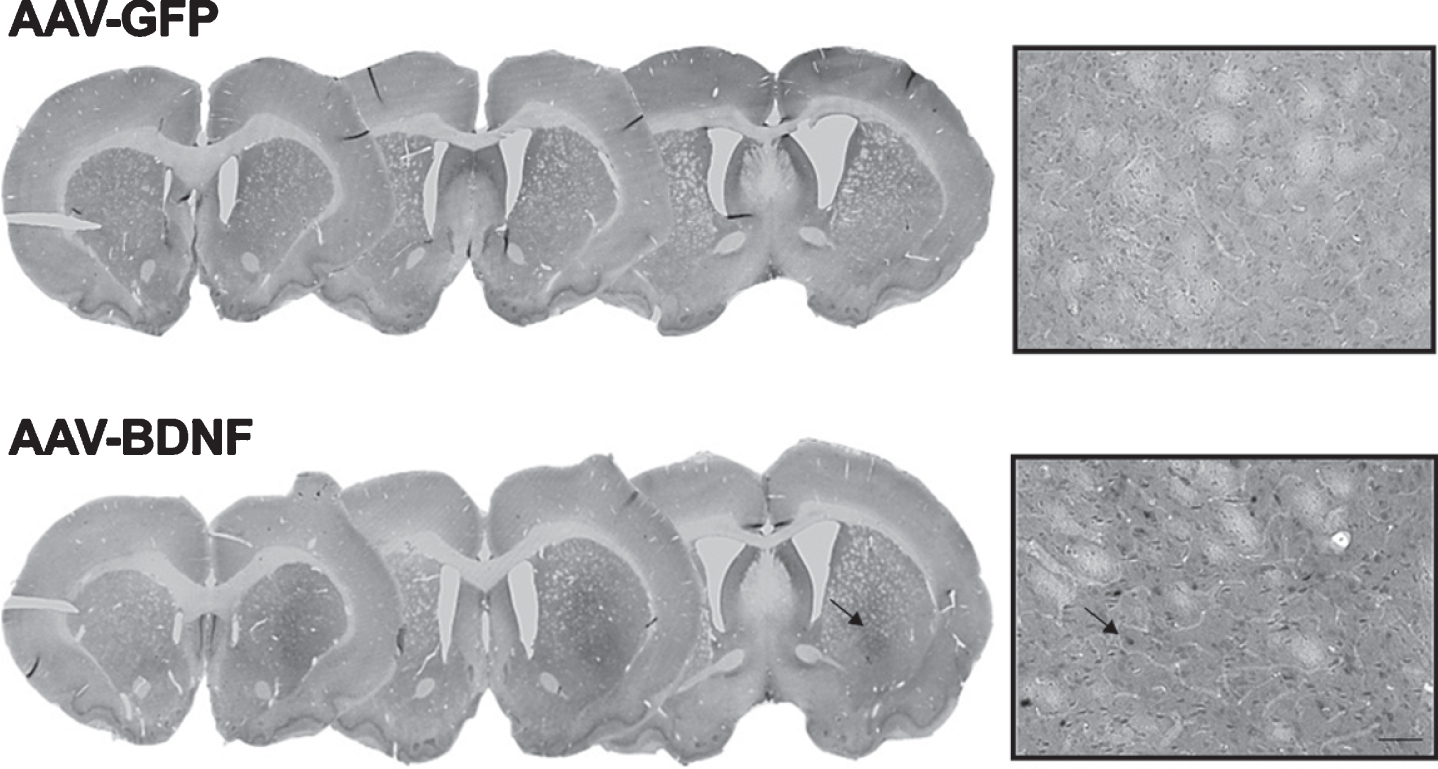
Representative photomicrographs of BDNF overexpression in 6-OHDA-lesioned rats unilaterally injected with the AAV vector for BDNF 3% or GFP 5%. A) Striatal coronal sections; B) 10x magnification of striatal sections. Scale bar: 100 μm.
Assessment of abnormal involuntary movements (AIMs)
AIMs were evaluated according to the rat dyskinesia scale described in detail previously [10–12]. Behavioral experiments occurred between 11 : 00 a.m. and 5 : 00 p.m. during the light phase of the light-dark cycle. Briefly, rats were placed individually in transparent plastic cages without bedding material and scored every 20 min following drug injection for the entire time course of dyskinesias (120 min). AIMs were classified into three subtypes according to their topographic distribution as forelimb, orolingual and axial behaviors. The severity of each AIM subtype was assessed using scores from 0 to 4 (1: occasional, i.e., present less than 50% of the time; 2: frequent, i.e., present more than 50% of the time; 3: continuous, but interrupted by strong sensory stimuli; 4: continuous, not interrupted by strong sensory stimuli). AIMs were scored blindly and independently by two investigators.
Immunohistochemistry
Perfused brains (ice-cold 4% paraformaldehyde in phosphate buffered saline (PBS) were transferred to 25% sucrose in PBS for cryoprotection overnight, and sections of 40 μm were obtained using freezing microtome (Leica, Wetzlar, Germany). Free-floating immunohistochemistry was performed as previously described [13]. Briefly, sections were incubated at room temperature with the primary antibody, rabbit anti-TH (1 : 1000, Merck #AB152), rabbit anti-BDNF (1 : 1000, Santa Cruz sc-546), and rabbit anti-GFP (1 : 20000, Abcam AB13970). On the second day, sections were incubated for 1 h in a 1 : 200 dilution of specific biotinylated secondary antibody. After rinsing, sections were treated with avidin-biotin complex (ABC Elite Kit, Vector Laboratories C at #PK-6100, RRID:AB_2336827) in KPBS for 1 h. The color reaction was developed by incubation in 25 mg/ml 3,3’-diamminobenzidine and 0.005% H2O2. TH immunohistochemistry was performed to verify the extent of dopaminergic lesion; all rats included in the study (severely impaired in the stepping test) were found to have a complete lesion of the nigrostriatal pathway (see Supplementary Figure 1B for representative staining).
Biochemical studies
Sample preparation
Right and left striata were excised using the rapid head-freeze dissection technique as previously described and both lesioned and intact sides were analyzed for each animal [14]. Frozen tissues were solubilized in cell lysis buffer (50 mM TRIS, pH 7.4, 250 mM NaCl, 5 mM EDTA, 50 mM NaF, 1 mM sodium orthovanadate, 1% Triton X-100, 0.02% NaN3) containing 1 mM phenylmethylsulfonyl fluoride and protease inhibitor cocktail (#P 8340, Sigma-Aldrich). For the analysis of phosphorylation levels of ERK1/2 and DARPP-32, lysates were centrifuged at 14,000×g at 4°C for 10 min. Protein content was determined by Lowry method.
Immunoblotting
Equal amount of protein (30 μg) were separated on a 4–15% TGX Stainfree Criterion precast gel (Bio-Rad Laboratories, Inc., Hercules, CA; #5678085) and transferred to a nitrocellulose membrane at 12V (constant) for 1 h [15,16, 15,16]. The membranes were incubated with primary antibodies against D1R and D3R (Santa Cruz Biotechnology #sc-33660, RRID:AB_668813, 1 : 1000 and Santa Cruz Biotechnology #sc-9114, RRID:AB_639197, 1 : 500), phospho p44/42 MAPK (p-ERK1/2; Thr202/Tyr204, Cell Signaling Technology #4370, RRID:AB_2315112, 1 : 1000), p44/42 MAPK (ERK1/2, Cell Signaling Technology #4695, RRID:AB_390779, 1 : 1000), phospho-Thr34 DARPP-32 (Cell Signaling Technology #12438, RRID:AB_2797914, 1 : 1000) and DARPP-32 (Cell Signaling Technology #2306, RRID:AB_823479, 1 : 1000), and BDNF (ab226843, Abcam; 1 : 2000) in blocking buffer overnight at 4°C. Specific antibody binding was detected by chemiluminescence with the ChemiDoc XRS + Imager (Bio-Rad Laboratories). Samples from rats in the four different experimental group were immunoblotted and analyzed together. To control for equal loading for D1R, D3R, BDNF, ERK1/2, DARPP-32 expression, blots incubated with antibodies were stripped and reprobed using anti β-actin (#A1978, Sigma-Aldrich, St. Louis, MO, USA); for phospho-ERK1/2 and phospho-Thr34 DARPP-32, blots were stripped and reprobed using anti total ERK or anti total DARPP-32 antibody, respectively. Samples from rats in the four experimental groups were run on the same immunoblots and then analyzed together. Bands were quantified in arbitrary units and normalized using the software Image Lab (Bio-Rad Laboratories) using β-actin, ERK1/2 and DARPP-32 as loading controls. Expression values expressed as arbitrary units were then calculated as percentage of the control (AAV–GFP + Saline) group values.
Immunoprecipitation
Immunoprecipitation was performed as previously described [16,17, 16,17]. Briefly, D1R were immunoprecipitated from tissue lysates using monoclonal D1R antibody against the last 123 C-terminal amino acids of rat D1R (SC-33660, Santa Cruz Biotechnology). Antibodies were coupled to protein A Dynabeads (#1004, Invitrogen) using 5 μg antibody by rotating the mixture for 10 min at room temperature. Beads were washed twice in PBS and then the antibody-conjugated beads were incubated with 200–300 μg of protein lysate for 2 h at 4°C, followed by 3 washing steps in 0.1% Tween 20 supplemented PBS. Bound proteins were then eluted with 4×XT Sample buffer and Reducing agent (Bio-Rad Laboratories). The immunoprecipitates were separated on a 4–15% TGX Criterion precast gel [17]. The membranes were incubated with polyclonal anti-D3R antibody (SC-9114, Santa Cruz Biotechnology). Chemiluminescence was detected and quantified with the ChemiDoc Imaging System (Bio-Rad Laboratories). Samples from rats in the four experimental groups were run on the same immunoblots and then analyzed together. Band intensities of co-immunoprecipitated D3Rs were normalized to the band intensities of immunoprecipitated D1Rs. Band intensities were quantified using Image Lab software/Gel Doc XRS + system; values, expressed as arbitrary units, were then calculated as percentage of the control (AAV–GFP+Saline) group values.
Statistical analysis
Statistical analysis was performed using Prism 7 Software (San Diego, CA, USA). Normality and homoscedasticity were preliminarily verified using Kolmogorov-Smirnov’s test. Behavioral data were analyzed by two-way Repeated Measure (RM) ANOVA with BDNF and time as factors; neurochemical data were analyzed by two-way ANOVA with BDNF expression and SKF82958 treatment as factors, followed by Bonferroni’s multiple comparison test, as appropriate. Correlation between neurochemical and behavioral parameters was performed using Pearson’s correlation test. Group size was determined by power analysis calculated using the variance estimates obtained from previous results in our labs [1, 16]. Alpha value was set at p < 0.05. All values are presented as mean±SEM.
RESULTS
Effect of BDNF-overexpression on SKF82958-induced dyskinesia
6-OHDA-lesioned rats with striatal overexpression of either BDNF or GFP (n = 9/group) were treated daily with a low dose of the selective D1R agonist SKF82958 (0.02 mg/kg/day s.c.), and tested for development of AIMs. SKF82958 induced significant exacerbation of AIMs in BDNF-overexpressing animals, compared to the GFP controls, at every time point (Fig. 1; two-way ANOVA for repeated measures –Time: F3,51 28.18, p < 0.0001; Treatment: F1,17 = 114.6, p < 0.0001; Time × Treatment interaction: F3,51 = 1.76, p = 0.17; post-hoc Bonferroni’s multiple comparison test: p < 0.0001 vs the corresponding time point of AAV–GFP+SKF82958 group). Twenty-four h after the behavioral analysis, animals were sacrificed in order to verify BDNF and TH expression by immunohistochemistry (Fig. 2 and Supplementary Figure 1B, respectively).
BDNF induced an increase in striatal dopamine D3 receptor levels in hemiparkinsonian rats
Different groups of BDNF and GFP-overexpressing rats were administered saline or SKF82958 (0.02 mg/kg/day s.c.) for 4 days and sacrificed at day 4, 20 min after the last SKF82958 treatment, for biochemical measurements of BDNF, D1R, D3R, and D1R–D3R co-immunoprecipitation, as well as phospho-Thr34 DARPP-32 and phospho-ERK1/2 levels.
A significant difference in the AIMs score between BDNF and GFP animals was observed also in this group (Supplementary Figure 2; two-way ANOVA for repeated measures –Time: F1,16 = 148.5, p < 0.0001; Treatment: F1,16 = 93.9, p < 0.0001; Time×Treatment interaction: F1,16 = 2.209, p = 0.1566; post-hoc Bonferroni’s multiple comparison test: p < 0.0001 vs the corresponding time point of AAV–GFP + SKF group).
First, we evaluated BDNF levels in order to verify the efficiency of AAV striatal injection. The results showed that local injection of the BDNF–AAV vector resulted in increased expression of mature BDNF in the striatum of our hemiparkinsonian rats (two-way ANOVA, BDNF overexpression: F1,20 = 69.90, p < 0.001; SKF82958 treatment: F1,20 = 0.823, p = 0.374; interaction: F1,20 = 0.240, p = 0.629; Fig. 3A).
Next, we questioned whether BDNF overexpression might have modified the total levels of expression of D3R. Results showed that striatal injection of the BDNF-overexpressing vector increased dopamine D3R expression levels compared to the GFP vector, while SKF82958 treatment had no further effect (two-way ANOVA, BDNF overexpression: F1,20 = 6.152, p = 0.0221; SKF82958 treatment: F1,20 = 0.556, p = 0.46; interaction: F1,20 = 0.0015, p = 0.969, Fig. 3B). Thus, BDNF overexpression increased total expression of D3R even in absence of pharmacological treatment. By contrast, dopamine D1R expression levels were not modified by these manipulations (two-way ANOVA, BDNF overexpression: F1,20 = 0.22, p = 0.64; SKF82958 treatment: F1,20 = 0.566, p = 0.46; interaction: F1,20 = 0.063, p = 0.803; Fig. 3C). Uncropped images of immunoblots are shown in Supplementary Figure 3.
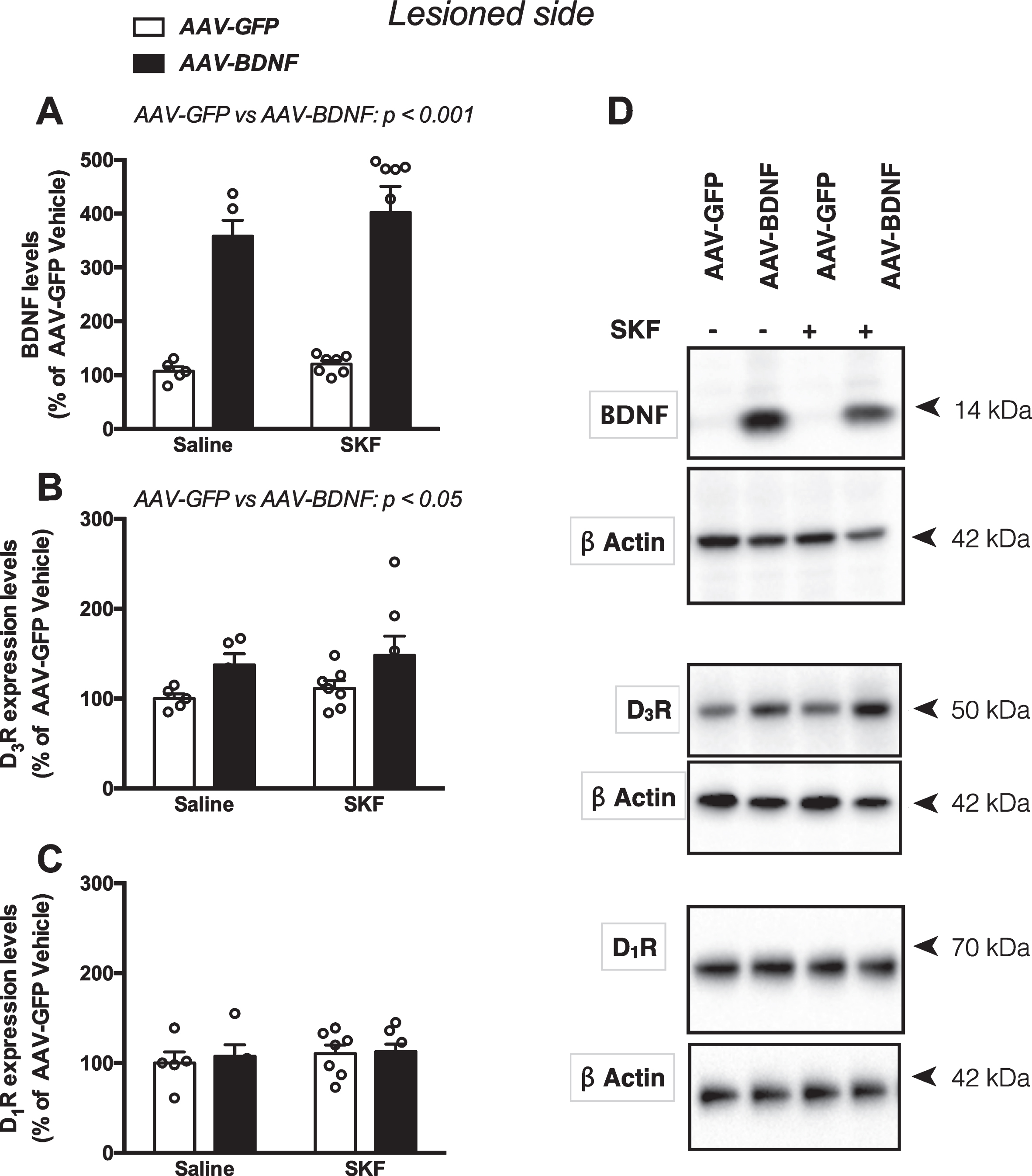
BDNF, D3R and D1R expression in BDNF and GFP overexpressing hemiparkinsonian rats. A) Levels of BDNF; B) levels of dopamine D3 receptors (D3R); C) levels of dopamine D1 receptors (D1R); D) representative immunoblots. Data are expressed as mean±SEM and calculated as percentage of AAV–GFP Saline values; n = 5–7/group; two-way ANOVA.
SKF82958 treatment increased D1–D3R co-immunoprecipitates in the striata of BDNF-overexpressing rats
Since a key role of dopamine D1R–D3R complexes has been demonstrated in the expression of LID [16, 18], we hypothesized that D3R would interact with D1R upon pharmacological treatment with the D1R agonist. Thus, we investigated the influence of BDNF overexpression and SKF82958 treatment on co-immunoprecipitation of D1R and D3R using an immunoprecipitation assay. The analysis of D1R–D3R levels in the immunoprecipitates by two-way ANOVA showed a significant effect of BDNF overexpression, SKF treatment and their interaction (AAV–BDNF F1,23 = 5.73, p = 0.025, SKF treatment F1,23 = 16.40, p = 0.0005 and their interaction F1,23 = 5.73, p = 0.0393; AAV–GFP Saline and AAV–BDNF Saline, AAV–GFP SKF, AAV–BDNF SKF). In particular, post-hoc analysis showed that D1R–D3R levels in the immunoprecipitates were significantly increased in animals overexpressing BDNF and treated with SKF82958 compared to saline treated animals overexpressing BDNF, or GFP-overexpressing animals treated with SKF82958 (p < 0.01 for both comparisons, Fig. 4A, B), thus indicating that BDNF overexpression per se was not sufficient to increase D1R–D3R immunoprecipitated formation. A significant correlation between D1R–D3R levels and AIMs score was found in SKF82958 treated animals overexpressing GFP or BDNF (AAV–GFP SKF, AAV–BDNF SKF groups) (Fig. 4C), further supporting the role of D1R–D3R interaction in the expression of dyskinesia [18]. By contrast, no significant correlation between D3R levels and AIMs score was found in SKF82958 treated animals overexpressing GFP or BDNF (AAV–GFP SKF, AAV–BDNF SKF groups); Fig. 4D.
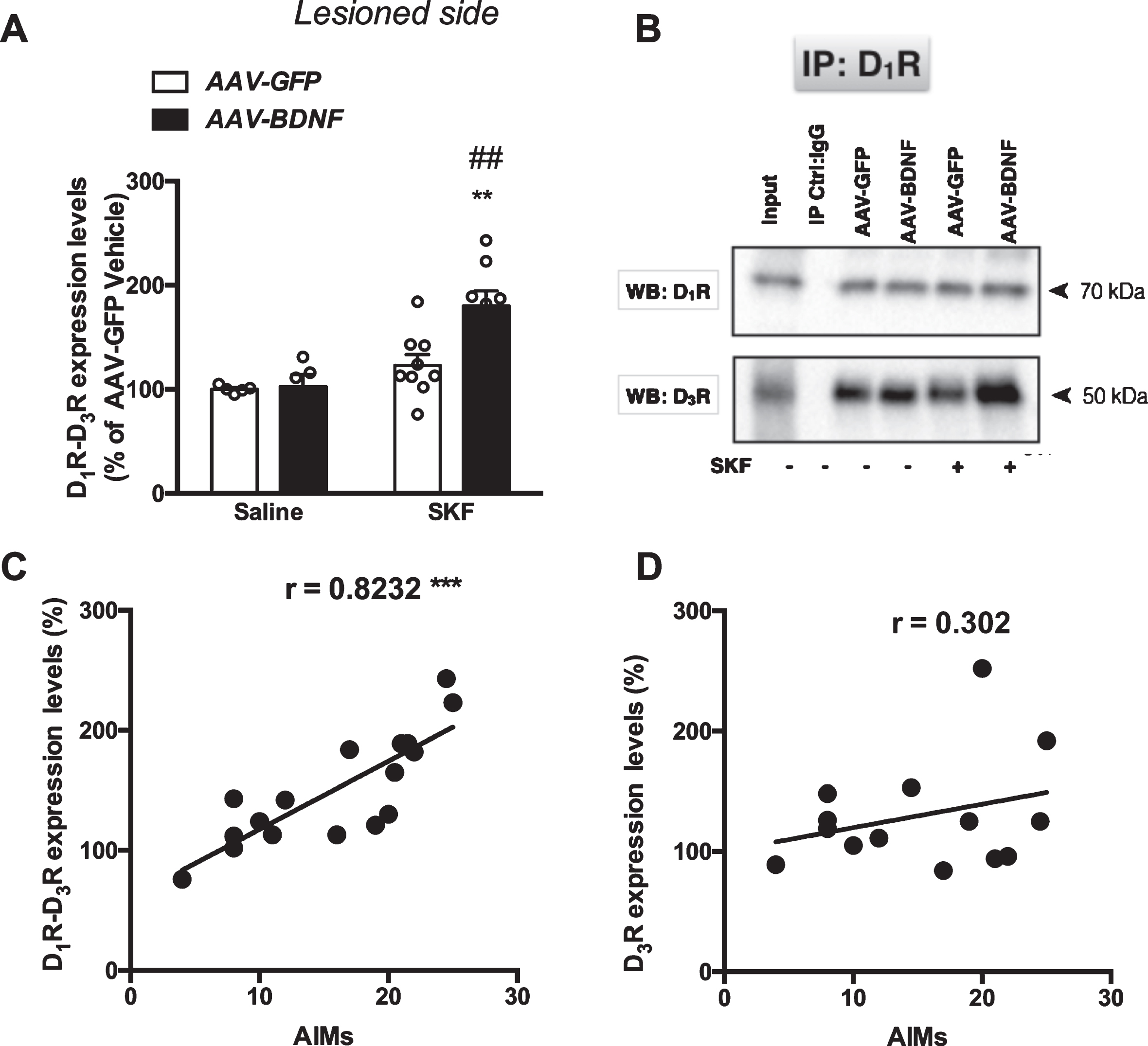
SKF82958 administration increased levels of D1–D3R complexes in the lesioned striatum of BDNF-overexpressing hemiparkinsonian rats. A) levels of D1–D3 receptor heteromers (D1–D3R); B) representative immunoblots. C, D) Correlation between the expression levels of D1–D3R (C) and D3R (D) and AIMs score in SKF82958-treated rats (AAV–GFP and AAV–BDNF groups). Tissue samples were immunoprecipitated with anti-D1R antibodies and immunoblotted using anti-D3R antibodies. Band intensities of co-immunoprecipitated D3Rs were normalized to the band intensities of immunoprecipitated D1Rs. Total striatal lysate without Co-IP (input) served as a positive control whereas normal mouse IgG was used for IP and served as a negative control. Data are expressed as mean±SEM and calculated as percentage of AAV–GFP Saline values. Two-way ANOVA followed by Bonferroni’s test; AAV–GFP Saline and AAV–BDNF Saline n = 5/group, AAV–GFP SKF n = 9/group, AAV–BDNF SKF n = 8/group; **p < 0.01 in comparison to AAV–BDNF Saline values; # # p < 0.01 in comparison to AAV–GFP SKF group (Bonferroni’s test). Note that for correlation studies both AAV–GFP + SKF and AAV–BDNF + SKF groups are plotted.
SKF82958 administration resulted in increased phosphorylation of Thr34 DARPP-32 and ERK1/2 in the striata of BDNF-overexpressing rats
To investigate whether the enhanced susceptibility to develop dyskinesia and the increased D1R–D3R co-immunoprecipitation induced by SKF82958 administration in rats overexpressing BDNF was also accompanied by alterations in dopamine D1R signaling, we analyzed phospho-Thr34 DARPP-32 and phospho-ERK1/2 levels, that can be regarded as striatal markers of dyskinesia [19]. The analysis of phospho-Thr34 DARPP-32 levels by two-way ANOVA showed a significant effect of BDNF overexpression (F1,23 = 14.30, p = 0.001), BDNF and SKF82958 interaction (F1,23 = 10.56, p = 0.0035), but not of SKF82958 administration (F1,23 = 3.628, p = 0.0694). In fact, SKF82958 treatment, at the dose used, increased phospho-Thr34 DARPP-32 levels only in animals overexpressing BDNF and showing the dyskinetic phenotype (AAV–GFP Saline and AAV–BDNF Saline, AAV–GFP SKF, AAV–BDNF SKF; Fig. 5A). The analysis of phospho-ERK1/2 levels by two-way ANOVA showed a significant effect of BDNF overexpression (F1,23 = 7.24, p = 0.0130), SKF82958 administration (F1,23 = 10.41, p = 0.0037), but not their interaction (F1,23 = 0.59, p = 0.4495; AAV–GFP Saline and AAV–BDNF Saline, AAV–GFP SKF, AAV–BDNF SKF; Fig. 5B).
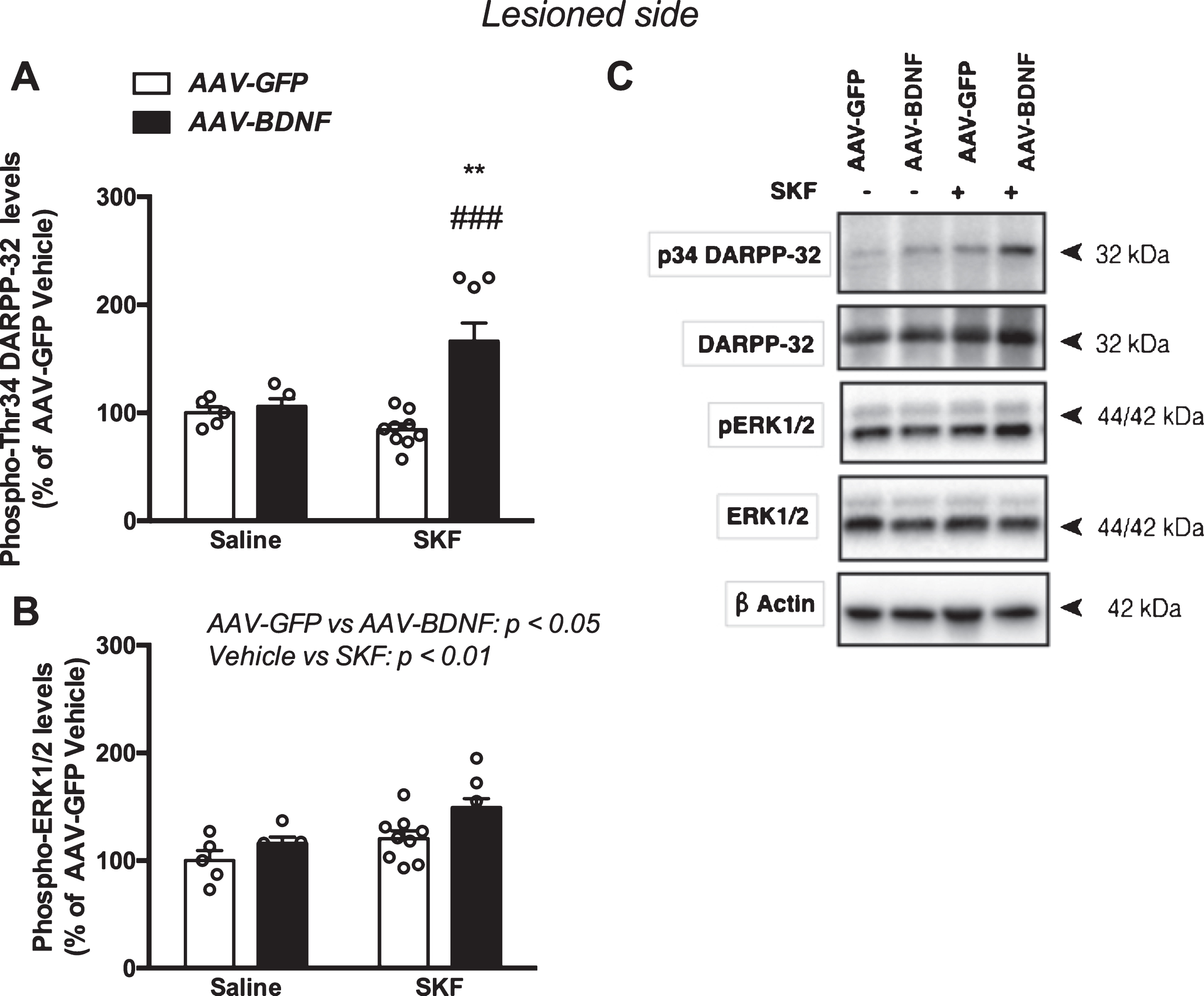
SKF82958 administration increased phosphorylation levels of Thr34 DARPP-32 and ERK1/2 in the lesioned striatum of BDNF-overexpressing hemiparkinsonian rats. A) Levels of phospho-Thr34 DARPP-32; B) levels of phospho-ERK 1/2; C) representative immunoblots. Phospho-Thr34 DARPP-32 and phospho-ERK1/2 levels were normalized to those of DARPP-32 and ERK1/2, respectively. Data are expressed as mean±SEM and calculated as percentage of AAV–GFP Saline values. Two-way ANOVA followed by Bonferroni’s test; AAV–GFP Saline and AAV–BDNF Saline n = 5/group, AAV–GFP SKF n = 9/group, AAV–BDNF SKF n = 8/group; **p < 0.001 in comparison to AAV–BDNF Saline values; # # # p < 0.001 in comparison to AAV–GFP SKF group (Bonferroni’s test).
Differently from the lesioned side, where the GFP or BDNF AAV vector was injected, in the intact side similar levels of D1R (Supplementary Figure 4A; two-way ANOVA, BDNF overexpression: F1,16 = 2.377, p = 0.144; SKF82958 treatment: F1,16 = 1.41, p = 0.252; interaction: F1,16 = 0.0038, p = 0.9847) and D3R (Supplementary Figure 4B; two-way ANOVA, BDNF overexpression: F1,16 = 0.897, p = 0.357; SKF82958 treatment: F1,16 = 0.0898, p = 0.768; interaction: F1,16 = 0.0138, p = 0.715), and D1R–D3R complexes (Supplementary Figure 4C; two-way ANOVA, BDNF overexpression: F1,16 = 1.525, p = 0.234; SKF82958 treatment: F1,16 = 2.845, p = 0.111; interaction: F1,16 = 1.452, p = 0.245) were observed in the immunoprecipitates. Moreover, in the intact side, phospho-ERK1/2 (Supplementary Figure 4D; two-way ANOVA, BDNF overexpression: F1,16 = 0.0287, p = 0.867; SKF82958 treatment: F1,16 = 0.131, p = 0.721; interaction: F1,16 = 1.646, p = 0.217) and phospho-Thr34 DARPP-32 levels (Supplementary Figure 4E; two-way ANOVA, BDNF overexpression: F1,16 = 0.888, p = 0.360; SKF82958 treatment: F1,16 = 0.311, p = 0.584; interaction: F1,16 = 0.030, p = 0.864) were similar between groups.
DISCUSSION
Here we show that viral vector overexpression of BDNF upregulated D3R levels in the striatum of 6-OHDA-lesioned rats, and that this event was associated with increased susceptibility to develop AIMs in response to D1R activation. The link between D1R and dyskinesia is well established [19–23]. Studies using D1R knockout mice showed that this receptor is critical for the development of dyskinesia and associated molecular markers [24, 25]. Moreover, pharmacological blockade of D1R reduces LID in different animal models [21, 27].
Previous research in animal models of PD has also linked the development of dyskinesia to the levels of expression of D3R. These receptors are usually sparsely expressed within the dorsal striatum [28]; however, they are significantly upregulated upon L-DOPA administration in dopamine-depleted animals, and they co-localize with D1R [29, 30]. Moreover, upregulation of D3R mRNA is induced by D1R agonist treatments, and L-DOPA-induced changes in D3R expression are attenuated by D1R antagonists, suggesting a possible interaction between these two receptors [29]. In situ proximity ligation assay showed that D1R and D3R co-localize within the striatum of dyskinetic animals [30], while previous co-immunoprecipitation and radioligand binding experiments suggest D1R–D3R heteromers formation [31, 32]. A key role of D3R in dyskinesia is also supported by the evidence that mice lacking D3R have reduced LID expression [33]. Furthermore, lentiviral-induced D3R overexpression in the mice dorsal striatum leads to the development of dyskinesia [34].
Thus, growing evidence suggests a functional cross-talk between D1 and D3 receptors in dyskinesia, and it has been postulated that D3R anchors D1R to the plasma membrane avoiding its internalization following receptor activation [35]. Under this conformation, D3R would increase D1R agonist affinity, allowing a stronger stimulatory coupling of the D1R to the cAMP signaling system and ERK1/2 pathway, and potentiating D1R-mediated behavioral responses [36, 37]. Accordingly, in the present study, repeated administration of a low dose of the D1R agonist SKF82958 was sufficient to increase co-immunoprecipitation of D1R–D3R, which has been suggested indicative of heteromers formation [31], and the phosphorylation levels of Thr34 DARPP-32 and ERK1/2 in the striatum of BDNF- but not GFP-overexpressing animals. Indeed, at the behavioral level, SKF82958 administration induced only sporadic dyskinetic movements in GFP- compared to BDNF-overexpressing rats. It is worth noting that BDNF overexpression per se did not modify the levels of striatal D1R–D3R complexes in our experimental setting, despite the increased availability of D3R. By contrast, D1R activation appears mandatory in order to trigger an increased D1R–D3R co-immunoprecipitation. It is possible that D1R activation is mandatory to recruit D3R at the synaptic membrane, and thus, form heteromers, although further investigations should be conducted to verify this hypothesis.
It is important to stress that here we used a very low dose of SKF82958 (0.02 mg/kg). This dose was specifically chosen to induce sporadic dyskinetic movements in the GFP-overexpressing group, in order to avoid the roof-effect in the dyskinesia rating scale that would have been induced by high doses, and, therefore, be able to fully appreciate the pro-dyskinetic effect of BDNF overexpression. This can explain the lack of effect of the D1R agonist administration on the expression of dyskinesia markers in the GFP-overexpressing animals, which is in apparent contrast with previous studies.
Whereas we induced here an artificial upregulation of striatal BDNF levels by using an AAV vector to overexpress this trophin, previous studies have indicated that BDNF can be induced by long term-treatment with L-DOPA in parkinsonian animals. For instance, Rylander and co-workers have shown that LID correlates with striatal levels of BDNF in 6-OHDA-lesioned rats [2]. This result is in agreement with other studies, which reported that behavioral sensitization to L-DOPA in 6-OHDA-lesioned rats was associated to upregulation of mRNA levels for BDNF and TrkB receptor in the frontal cortex and striatum [5, 29]. Therefore, while previous research has shown that BDNF and D3R may both play a key role in development of dyskinesia induced by long-term treatment with L-DOPA, the present investigation, using an AAV vector-mediated upregulation of striatal BDNF levels, points to the existence of a direct link between these two elements, which may play a detrimental role in dyskinesia. However, further research is needed to fully clarify the causal link between BDNF and D3R in dyskinesia.
Of note, altered spiny density/morphology and synaptic plasticity have been found in the striatum of parkinsonian rats [38, 39]. Interestingly, previous research showed that BDNF can promote long-term potentiation in striatal and hippocampal neurons, and increase hippocampal dendritic spine density [40, 41]. Moreover, striatal BDNF overexpression was shown to counteract the reduction of spine density and reverse abnormal spine morphology in striatal neurons of YAC128 mice [42]. Thus, it is conceivable to think that BDNF overexpression may also affect striatal synaptic plasticity and/or spine density/morphology in 6-OHDA-lesioned rats. In line with this hypothesis, the AIMs score was already increased upon the first SKF82958 administration in BDNF-overexpressing rats compared to GFP controls in the present study, suggesting that BDNF-induced alterations had developed in the lesioned striata independently from the D1R agonist administration. Further studies are needed to investigate this point.
We have previously shown that BDNF overexpression can affect the development of LID also by triggering sprouting of striatal serotonergic neurons [1]. Thus, taken together, our studies suggest that BDNF can affect dyskinesia by at least two mechanisms: first, by acting on the pre-synaptic compartment and providing an augmented buffering capacity for the exogenous L-DOPA, which would trigger increased dopamine production and pulsatile stimulation of dopamine receptors [1]; second, by acting on the post-synaptic compartment and potentiating the D1R signaling cascade, likely via increased D1R–D3R interaction. In this scenario, more dopamine would be released on supersensitive dopaminergic receptors, leading to stronger molecular alterations and increased susceptibility to dyskinesia.
In conclusion, we showed here that BDNF overexpression exacerbates dyskinesia induced by direct D1R agonists in 6-OHDA-lesioned rats, as previously seen with L-DOPA, and we provide evidence for a functional link between BDNF, D3R and D1–D3R close interaction in the augmented susceptibility to dyskinesia. We suggest that D1–D3 receptors interaction may be instrumental in driving the molecular alterations underlying the appearance of dyskinesia and represents a possible therapeutic target for treatment of this condition in PD patients. In fact, increased D3R binding has been found in dyskinetic subjects using positron emission tomography [43], showing that the contribution of D3R to dyskinesia is not limited to the animal models but extends to PD patients.
CONFLICT OF INTEREST STATEMENT
The authors have not conflict of interest to declare.
Footnotes
ACKNOWLEDGMENTS
We thank Pierluigi Saba, Barbara Tuveri, Jenny G. Johansson and Augusta Pisanu for their expert technical assistance. This work was supported by grants from the Italian Ministry of Education, Universities and Research, Progetto di Ricerca di Interesse Nazionale (PRIN) 2010-AHHP5H.