
Abstract
Mitochondrial dysfunction represents a well-established player in the pathogenesis of both monogenic and idiopathic Parkinson’s disease (PD). Initially originating from the observation that mitochondrial toxins cause PD, findings from genetic PD supported a contribution of mitochondrial dysfunction to the disease. Here, proteins encoded by the autosomal recessively inherited PD genes Parkin, PTEN-induced kinase 1 (PINK1), and DJ-1 are involved in mitochondrial pathways. Additional evidence for mitochondrial dysfunction stems from models of autosomal-dominant PD due to mutations in alpha-synuclein (SNCA) and leucine-rich repeat kinase 2 (LRRK2). Moreover, patients harboring alterations in mitochondrial polymerase gamma (POLG) often exhibit signs of parkinsonism. While some molecular studies suggest that mitochondrial dysfunction is a primary event in PD, others speculate that it is the result of impaired mitochondrial clearance. Most recent research even implicated damage-associated molecular patterns released from non-degraded mitochondria in neuroinflammatory processes in PD. Here, we summarize the manifold literature dealing with mitochondria in the context of PD. Moreover, in light of recent advances in the field of personalized medicine, patient stratification according to the degree of mitochondrial impairment followed by mitochondrial enhancement therapy may hold potential for at least a subset of genetic and idiopathic PD cases. Thus, in the second part of this review, we discuss therapeutic approaches targeting mitochondrial dysfunction with the aim to prevent or delay neurodegeneration in PD.
Keywords
INTRODUCTION
The prevalence of Parkinson’s disease (PD) has more than doubled over the last two decades, making it the fastest growing of all neurological diseases [1]. Despite significant advances in deciphering the pathophysiology of PD [2], the etiology remains elusive for the majority of cases.
On the cellular level, an involvement of oxidative stress, lysosomal and mitochondrial dysfunction has been implicated in the pathophysiology of PD [3]. The first evidence that alterations in mitochondrial function may play a decisive role in the pathogenesis of PD date back to the 1980s, when mitochondrial toxins were reported to cause dopa-responsive parkinsonism [4]. Subsequently, findings from PD genetics supported the link between mitochondria and PD [5]. Here, it has been shown that mutated genes causing monogenic PD encode proteins involved in mitochondrial function and degradation of damaged mitochondria. This review aims to 1) discuss the origin of the link between PD and mitochondria, 2) summarize how pathogenic variants in the PD genes Parkin, PTEN-induced kinase 1 (PINK1) and DJ-1 as well as parkinsonism-associated mutations in mitochondrial Polymerase gamma (POLG) cause mitochondrial impairment, and 3) present how oxidative stress leads to mitochondrial DNA (mtDNA) disintegration in PD. Moreover, 4) we illustrate how mitochondrial damage may cause inflammation in the context of PD. Additionally, 5) we summarize the interaction between mitochondrial and lysosomal pathways as well as the endoplasmic reticulum (ER) with a focus on calcium homeostasis. Finally, 6) we discuss resulting implications for genetic testing and highlight possible therapeutic approaches arising from a potential mitochondrial subtype of PD.
ORIGINS OF THE LINK BETWEEN MITOCHONDRIA AND PD
First, the so-called “frozen addicts” suggested a contribution of mitochondrial dysfunction to the pathogenesis of PD. In these drug users, living in California in the 1980s, physicians observed that a side product of new synthetic heroin triggered a rapid onset of a distinct form of parkinsonism responsive to levodopa treatment. It turned out that the synthesis process resulted in the unwanted generation of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP), which led to inhibition of the respiratory chain [4]. Of note, a similar observation was published already four years earlier [6]. MPTP is not toxic itself but lipophilic and thus able to enter brain tissue by crossing the blood brain barrier. In the brain, it is processed by monoamine oxidase B (MAO-B) [7] to the toxic cation 1-methyl-4-phenylpyridinium (MPP+) [8]. MPP+is selectively taken up by dopaminergic cells [9] and inhibits multiple complexes of the respiratory chain [3, 10]. The notion that mitochondrial dysfunction plays a role in PD pathogenesis was supported shortly after the description of the “frozen addicts” by the observation of a restricted function of respiratory chain complexes in postmortem brain sections from PD patients [11]. These early findings significantly stimulated PD research in the following years. For example, even today, the injection of MPTP is most commonly used to model PD in mice [12]. However, similar to other animal models of PD, the clinical and pathological characteristics simulated by the MPTP model differ from PD in many ways [13].
Disturbances in respiratory chain complexes are associated with the generation of reactive oxygen species (ROS) suggesting oxidative stress as a pathogenic mechanism in PD related to mitochondrial dysfunction. Highlighting the role of ROS, evidence has arisen that oxidative stress is linked to dopamine metabolism [14]. Later in the present review, we will particularly focus on the aspect of oxidative stress and mtDNA disintegration.
MONOGENIC PD AND MITOCHONDRIAL DYSFUNCTION
Over the past two decades, intensive research has resulted in significant progress regarding the elucidation of monogenic causes of PD. After the initial description of pathogenic variants in the alpha-synuclein gene (SNCA) as of cause PD in 1997 [15], several genes have been identified that are associated with the development of PD signs resembling those of idiopathic PD. These genetic alterations are considered as disease-causing or as genetic risk factors. In particular, the autosomal dominantly inherited genes SNCA, Leucine-rich repeat kinase 2 (LRRK2), and Vacuolar protein sorting-associated protein 35 (VPS35) [16] and the autosomal recessively transmitted genes Parkin, PINK1 and DJ-1 [17] are both well established and validated to cause PD when mutated. In addition, a number of genes have been shown to cause atypical parkinsonism [18].
In the context of autosomal dominantly inherited PD, several links to mitochondrial dysfunction have been described in the past decade. For instance, the protein encoded by the first PD-linked gene SNCA is a component of Lewy bodies [19], which were recently also identified to contain organelles including mitochondria [20]. Alpha-synuclein has been shown to accumulate in mitochondria, interfering with complex I function and increasing mitophagy [21]. Thereby, calcium can trigger alpha-synuclein-mediated mitochondrial dysfunction [22, 23]. In keeping with these findings, the N-terminal domain of alpha-synuclein is associated with respiratory chain complex I [24]. Moreover, neuroepithelial stem cells (NESCs) harboring PD-causing SNCA mutations showed reduced mitochondrial function [25]. In addition, a nonfibrillar, phosphorylated species of alpha-synuclein has been shown to target mitochondria, thereby inducing mitochondrial fragmentation, energy deprivation and mitophagy [26]. The role of alpha-synuclein at the mitochondria-associated endoplasmic membrane (MAM) will be discussed below in a separate section on inter-organellar crosstalk.
There is also evidence for a role of LRRK2 in the regulation of mitochondrial function. Mutations in LRRK2 cause the most common and autosomal dominantly inherited form of monogenic PD clinically indistinguishable from IPD [27, 28]. As described later in this review, Parkin and PINK1 play a well-established role in a common pathway mediating mitophagy, the process of degrading damaged mitochondria. Similarly, LRRK2 is involved in the initiation of mitophagy by regulating mitochondrial motility [3]. Further evidence for an involvement of LRRK2 in mitochondrial clearance comes from our own observation of elevated mtDNA deletion levels specifically in affected LRRK2 mutation carriers, implicating mtDNA integrity as potential penetrance marker for LRRK2-linked PD [29]. Concerning mutations in VPS35, another cause of autosomal dominantly inherited PD [30], there is also evidence for an association with mitochondrial dysfunction. For example, VPS35-mutant fibroblasts exhibited an impaired configuration of complex I of the respiratory chain [31]. In dopaminergic neurons, VPS35 depletion leads to the accumulation of α-synuclein and mitochondrial dysfunction [32]. An additional mechanistic link between VPS35 and mitochondria was demonstrated when the fission factor dynamin-like protein (DLP) 1 emerged as interactor of VPS35 [33].
Moreover, the PD-associated protein CHCHD2 [34] has been found to accumulate in mitochondria under the influence of stress [35]. Further studies will be needed to shed light on its interaction with CHCHD10 [36].
However, the most compelling evidence for a direct link between mitochondria and PD has been established for the autosomal recessively inherited PD genes Parkin, PINK1, and DJ-1, as illustrated by a PubMed search: Combining “Parkinson’s disease AND mitochondria” with any of these three gene names results in over 4500 publications in total. Interestingly, patients with genetic alterations in the mitochondrial disease-associated gene POLG also exhibit parkinsonism, albeit a clinically more atypical form.
Parkin-linked PD
Clinically, biallelic mutations in Parkin cause typical levodopa-responsive PD with early disease onset, slow progression and dystonia as prominent (initial) symptom, while non-motor features like olfactory dysfunction, psychiatric symptoms or cognitive impairment are less frequent compared to IPD [17] (Table 1).
Overview of genes particularly associated with mitochondrial dysfunction in Parkinson’s disease and POLG as representative of genetic mitochondrial disease with parkinsonian features
*Taken from www.mdsgene.org; table according to [17, and 145]; mtDNA, mitochondrial DNA; MDS, Movement Disorder Society; OMIM, Online Mendelian Inheritance in Man; PINK1, PTEN-induced kinase 1; POLG, Polymerase gamma.
In 1997, an unidentified gene mapping to chromosome 6q25.2–27 was initially linked to an autosomal recessive juvenile form of parkinsonism [37]. Shortly thereafter, the sequence of Parkin was unveiled, with subsequent reports furthering its significance for the etiology of PD [38]. To date, more than 130 different mutations in Parkin have been documented in about 1000 PD patients [17], making it the most prevalent autosomal recessive form of PD [39]. Parkin is an E3 ubiquitin ligase with established neuroprotective activities. Furthermore, Parkin has an extensive array of putative substrates [40], which can be differentially modified either through mono- or poly-ubiquitination with different patterns of ubiquitin lysine linkage. This results in a complex, yet insufficiently characterized array of regulatory nodes associated to this protein. Parkin exerts its function through three independent mechanistic axes [41]: 1) enhanced ubiquitination of toxic substrates to be degraded by the proteasome, 2) regulation of cell death pathways through non-degradative ubiquitin signaling, and 3) regulation of mitochondrial quality control through mitophagy and vesicular transport. Although initial reports failed to detect mitochondrial localization of Parkin [42], it is currently established that this protein is intimately related to the regulation of mitochondrial homeostasis.
Lys-48-polyubiquitinated Parkin substrates are directed to the proteasomal degradation pathway [43], meaning that Parkin deficiency or inactivation can lead to accumulation of diverse noxious substrates that are normally targeted for degradation. A good example of this is PARIS, a repressor of the master regulator of mitochondrial biogenesis and respiration, PGC1-α [44], as will be further explained below. The first indisputable evidence for parkin’s involvement in mitochondrial homeostasis arose from the study of Drosophila [45] and mouse [46] parkin–/– models. Remarkably, these fly models exhibited degenerative phenotypes, which considerably overlapped with those reported soon thereafter in pink1–/– fly models [47–49], exposing a mechanistic link between parkin, pink1 and mitochondrial quality control processes which will be further addressed below.
PINK1-linked PD
Autosomal recessively inherited mutations in PINK1 cause early-onset PD with similar clinical features as described for PD due to biallelic Parkin mutations [17]. However, non-motor symptoms are slightly more frequent in PINK1- compared to Parkin-linked PD [17] (Table 1).
In 2001, a seminal study identified a novel locus for autosomal recessive early-onset parkinsonism at chromosome 1p35–p36 [50], which would later prove to be PINK1 [51]. PINK1 encodes a serine/threonine kinase possessing a mitochondrial translocation sequence, which led to the recognition of the protein’s involvement in mitochondrial function [51]. The kinase activity of PINK1 has been shown to be regulated by autophosphorylation on specific sites within the kinase domain (Ser228, Ser402 and Thr257) [52–54]—a process which is, for example, essential for Parkin translocation to the mitochondria upon mitochondrial stress [53] (Fig. 1).
In 2006, a series of reports on pink1-deficient Drosophila models exposed the interaction between pink1 and parkin [47–49]. Pink1-deficient male flies were sterile, exhibited marked degeneration of flight muscles and of dopaminergic neurons, and displayed altered mitochondrial ultrastructure that evidenced malfunction [47–49]. Strikingly, these pink1-related phenotypes were consistently replicated in parkin-deficient flies and could be reversed by overexpression of parkin in pink1-deficient flies, but not the inverse. These studies set the stage for the elucidation of the molecular regulatory pathway through which PINK1 and Parkin jointly act to warrant mitochondrial quality control. The predominant model suggests that PINK1 is constitutively expressed and translocated to mitochondria [51], where it functions as a sensor and tag for mitochondrial depolarization and malfunction [55–57]. Under steady-state conditions, PINK1 is readily imported into mitochondria through the TOM/TIM complex, whereby it is processed by the mitochondrial processing peptidase and cleaved by the PARL protease. The released N-terminal-deleted PINK1 fragment is ubiquitinated and degraded by the proteasome [56]. However, under dysfunctional conditions, such as loss of the mitochondrial membrane potential, this processing of PINK1 is inhibited [55, 58], resulting in its stabilization on the outer mitochondrial membrane where it phosphorylates diverse substrates (Fig. 1). Relevant at this level is the phosphorylation of ubiquitin Ser65 and, particularly, the direct phosphorylation of Parkin on Ser65 in its ubiquitin like domain, which has an allosteric effect [43]. This results in the recruitment and activation of Parkin and initiates the complex process of selective removal of damaged mitochondria through mitophagy [55], which has been thoroughly explained elsewhere [56]. Of note, mutations in the PD-linked kinase LRRK2 interfere with Parkin/PINK1-mediated mitophagy in a kinase activity-dependent manner [59] (Fig. 1). Further linking LRRK2 mutations and impaired mitophagy, a recent study demonstrated a Parkin and PINK1-dependent accumulation of RAB10 [60].
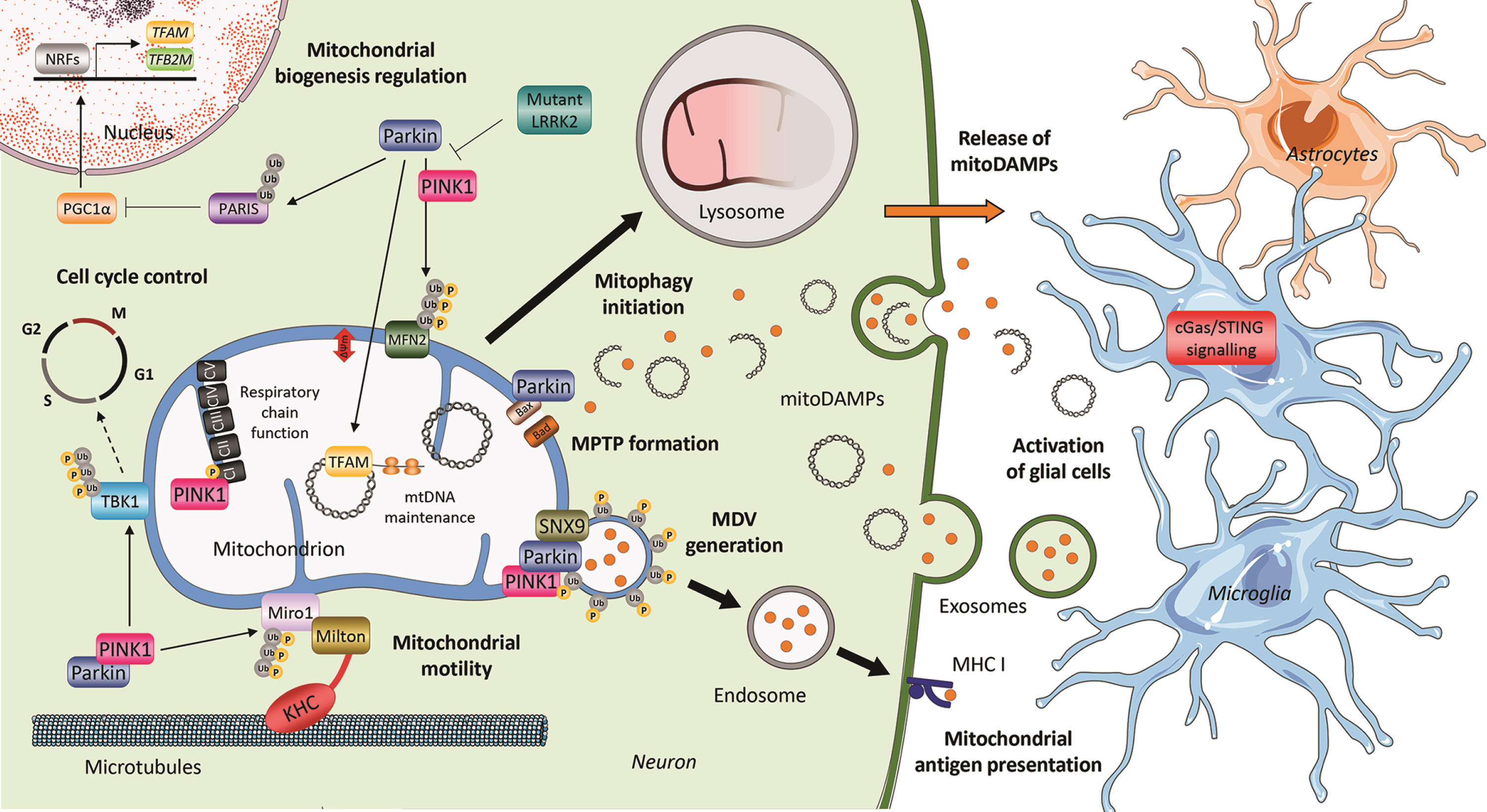
Involvement of PINK1 and Parkin in mitochondrial processes. The most investigated function of PTEN-induced putative kinase 1 (PINK1) and Parkin is the initiation of mitophagy. A loss in membrane potential triggers the PINK1-mediated recruitment of the E3 ubiquitin ligase Parkin to mitochondria. At the outer mitochondrial membrane, Parkin ubiquitinates proteins thereby tagging dysfunctional mitochondria for lysosomal degradation. This process can be inhibited by mutant LRRK2. In addition, both PINK1 and Parkin, in conjunction with Snx9, are involved in the formation of mitochondria-derived vesicles (MDVs), which can transport cargo such as mitochondrial damage-associated molecular patterns (mitoDAMPs). After engulfment of MDVs by endosomes, mitochondrial antigens are transported to the plasma membrane, where they are presented on histocompatibility complex class I (MHC I) molecules. MitoDAMPs can also be released from mitochondria trough the mitochondrial permeability transition pore (MPTP), which is formed under the control of Parkin –an interaction partner of the pro-apoptotic protein BCL2-antagonist/killer (BAK). In a PINK1- or Parkin-deficient environment, mitoDAMPs accumulate extracellularly and trigger cyclic GMP-AMP synthase/stimulator of interferon genes (cGas/STING) inflammatory signaling. However, the exact release mechanisms of mitoDAMPs and their impact on the interplay of neuronal and glial cells remain to be studied in human-derived PD models. In addition to its role in mitophagy, Parkin can modulate mitochondrial biogenesis by ubiquitination of the Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1-α) inhibitor PARIS or by direct interaction with the mitochondrial transcription factor A (TFAM) at the mtDNA. Moreover, Parkin influences cell cycle progression via its ubiquitination target TANK-binding kinase 1 (TBK1). By controlling the degradation of the microtubule adaptor protein Miro1, which links kinesin heavy chain (KHC) to mitochondria, PINK1 and Parkin regulate mitochondrial arrest as a prerequisite for mitochondrial clearance. Finally, there is also evidence for a direct interaction between PINK1 and respiratory chain complex I. Accordingly, PINK1 influences the activity of complex I by phosphorylation of its subunit NADH:ubiquinone oxidoreductase subunit A10 (NdufA10). The online image library Servier Medical Art (http://smart.servier.com/) was used to create this Figure, which is partially based on our previous review [3].
Besides mitophagy, the mitochondrial quality control program encompasses other mechanisms for the specific removal of localized damaged mitochondrial components. This is accomplished by means of mitochondrial-derived vesicles (MDVs), a particular type of vesicular trafficking [61]. MDVs can be generated as a response to stress [62], and can incorporate damaged cargo such as oxidized proteins which might then be eliminated through lysosomal degradation [3, 61]. Here again PINK1 and Parkin seem to serve as instrumental factors for the formation of MDVs [63] (Fig. 1). Moreover, the outer mitochondrial membrane protein Miro1, which links mitochondria to microtubule motor proteins during transport, is also a target of the Parkin/PINK1 pathway. Miro1 is degraded during the early stages of mitophagy thereby preventing further movement of dysfunctional mitochondria [64] (Fig. 1). In addition, Miro1 was shown to interact with LRRK2, a function that is hampered by the presence of pathogenic mutations, leading to reduced mitophagy and neurodegeneration [65].
The mechanisms through which PINK1 regulates mitochondrial homeostasis are not restricted to the aforementioned quality control process. Under steady-state conditions, PINK1 patient-derived fibroblasts and neurons display diminished complex I activity. This dysfunction was correlated to a specific loss of phosphorylation of serine-250 in the complex I subunit NdufA10 secondary to PINK1 deficiency [66] (Fig. 1). This is a good example of the complex and multifaceted regulatory process exerted by PINK1, and exposes its diverse range of actions under steady-state and stress conditions.
Although mitophagy represents a well-established mechanism in Parkin/PINK1-dependent PD, evidence for its role in PD in general is limited. Decreased mitophagy was demonstrated in IPD in a few studies on IPD fibroblasts and induced pluripotent stem cell (iPSC)-derived neurons [3]; however, the majority of results concerning genetic PD still stem from overexpression models [67]. Thus, the endogenous role of Parkin and PINK1 will require further investigation. Moreover, it is currently unknown how the genetic lack of these proteins specifically causes dopaminergic neurodegeneration. Given the ubiquitous expression of Parkin and PINK1 throughout the body, the absence of more wide-spread pathology also remains puzzling. These important research questions should be addressed in future studies.
DJ-1-linked PD
Mutations in the gene encoding the protein deglycase DJ-1 cause autosomal recessive PD [68] (Table 1), but are less common than mutations in Parkin or PINK1. Regarding DJ-1, several mechanistic links to impaired mitochondrial function have been described. First, the absence of DJ-1 alters mitochondrial morphology [69]. Moreover, in line with the already mentioned role as ROS scavenger in PD, an association between dopamine oxidation, mitochondrial, and lysosomal dysfunction was demonstrated in iPSC-derived neurons with mutations or depletion of DJ-1 in human and mice, respectively [70]. In keeping with this finding, also alterations in respiratory chain complex integrity were described in DJ-1-depleted neuronal cells [71].
POLG-related parkinsonism
In 2001, a preliminary study reported an association between POLG mutations and progressive external ophthalmoplegia (PEO) in three different Belgian families [72]. Thereafter, POLG mutations have been linked to an extraordinarily large set of disorders comprising a mitochondrial component, such as Alpers-Huttenlocher syndrome and, remarkably, recessively and dominantly inherited parkinsonism [73–75]. Interestingly, rare polymorphic variants of POLG have been suggested to pose a risk factor for IPD [76–78]. As discussed in the following, this hypothesis is supported by the observation of enhanced somatic variability in the mitochondrial genome of IPD patients. POLG is the only known mammalian polymerase present in mitochondria, where it integrates the molecular complex responsible for mtDNA polymerization [79]. The functional complex is composed of a catalytic subunit encoded by the nuclear gene POLG and a homodimer accessory protein encoded by the POLG2 gene [75]. Adding to its polymerase activity, POLG additionally encompasses exonuclease function (which assures fidelity of mtDNA replication [80]) and 5’ deoxyribose phosphate lyase activity. The latter function is instrumental for the base excision repair process necessary to correct oxidative damage to mtDNA [79, 81]. Overall, the combination of these three enzymatic competencies place POLG as a key player in the maintenance of mtDNA homeostasis. Therefore, it is not surprising that mutations, which compromise POLG function can lead to mitochondria-associated disorders including parkinsonism. However, it is worth mentioning that POLG-associated Alpers disease does not represent the only mitochondrial disorder including parkinsonism in its clinical spectrum. For instance, parkinsonism in combination with PEO has also been reported in patients with mutations in TWNK [82, 83].
OXIDATIVE STRESS AND mtDNA DISINTEGRATION IN PD
As summarized in the previous sections, multiple lines of evidence point towards a role of oxidative stress in the pathogenesis of PD. In addition to toxin-induced or primary respiratory chain dysfunction, the auto-oxidation of dopamine can generate free radicals and active quinones [84]. These ROS have the capacity to damage the mitochondrial genome, causing single- and double-strand breaks [85]. The 16,569 bp-long circular mtDNA codes for few but critical subunits of the respiratory chain complexes I, III, IV, and V. When nicks in the mtDNA are repaired inefficiently, mtDNA point and deletion mutations develop [86]. To protect the mtDNA from oxidative insults, it is packaged in nucleoids by the mitochondrial transcription factor A (TFAM) [87]. By contrast, in dopaminergic neurons from IPD patients, TFAM deficiency has been observed [88, 89], suggesting an enhanced exposure of the mitochondrial genome to ROS.
Transmitochondrial cytoplasmic hybrid (or short cybrid) studies first implicated mtDNA alterations in the pathogenesis of PD. In these experiments, cybrids were created by fusing mature platelets (which naturally lack nuclei) from PD patients with mtDNA-depleted control cells. Introducing patient mtDNA into a control nuclear background sufficed to recapitulate PD-associated mitochondrial phenotypes in the receiving cells [3]. While there is currently no evidence to suggest a role for inherited mtDNA mutations in PD [3], somatic alterations in the mitochondrial genome are likely part of the disease process [90]. Investigating the mitochondrial genome in single postmortem substantia nigra neurons revealed mtDNA copy number depletion and an accumulation of major arc deletions in IPD patients [88, 92]. Moreover, polygenic risk score analyses of whole exome sequences from large IPD cohorts showed increased genetic variation in the mtDNA maintenance pathway [93].
With regard to genetic PD, an additional area of action of Parkin, besides the regulation of mitophagy, lies in the control of mitochondrial biogenesis. A series of studies in mice, drosophila and cell lines showed that the degradation of PARIS, a repressor of PPARGC1A expression, is mediated by Parkin. In this manner, Parkin controls the PGC-1α-induced transcription of nuclear-encoded mitochondrial proteins [44, 95]. However, this finding still awaits confirmation in endogenous PD patient-derived cells. In addition, there is evidence that Parkin’s mitochondrial biogenesis-modulating effect extends to the mitochondrial genome. As PGC-1α was identified as an interactor of the mitochondrial transcription factor A (TFAM) [96], Parkin could convey its action on the mitochondrial genome in an indirect fashion. In addition, in vivo and in vitro immunoprecipitation analyses identified a direct association of Parkin with the mitochondrial genome and TFAM [97, 98]. By binding to the transcription factor in the mitochondrial D-loop region, Parkin may catalyze (multiple) mono-ubiquitylation [99] of TFAM thereby modulating mtDNA gene expression. Further supporting an involvement of Parkin in mtDNA maintenance, crossing parkin knockout mice with “mutator” mice that harbor a proof reading-deficient version of mitochondrial polg revealed 1) an increase in pathogenic mtDNA mutations, 2) enhanced loss of nigral tyrosine hydroxylase-positive neurons, and 3) motor deficits in the double-mutant animals [100]. These results highlight the protective action of Parkin against mtDNA mutagenic stress —a role which is likely intertwined with the protein’s newly identified function in inflammatory signaling. Inflammation triggered by mitochondrial damage associated molecular patterns (DAMPs) as emerging topic in PD research will be discussed in more detail in the following section.
MITOCHONDRIAL DAMAGE-INDUCED INFLAMMATION IN PD
First results suggesting a link between TFAM shedding, mtDNA release and inflammation came from fundamental studies outside of PD research. In mouse embryonic fibroblasts (MEFs), a heterozygous tfam knockout was employed to genetically induce mtDNA stress [101]. Aberrant packaging of the mitochondrial genome due to tfam deficiency led to the escape of mtDNA from the mitochondria. In the cytosol, mtDNA can act as DAMP promoting cGAS/STING inflammatory signaling [101]. During apoptosis, mitochondrial DAMPs can be released through the mitochondrial permeability transition pore. The formation of BAK/BAX [102] or VDAC macropores [103] at the outer mitochondrial membrane has been shown to facilitate mitochondrial herniation and subsequent mtDNA efflux. Interestingly, the PD protein Parkin can ubiquitinate BAK thereby suppressing pore formation [104], cytochrome c release and consequent apoptosis induction [105, 106] to ensure efficient clearance of damaged mitochondria, which could otherwise trigger inflammation. A specific role for Parkin and PINK1 in mitochondrial damage-induced inflammation was further supported by a recent study in the above-mentioned parkin knockout “mutator” mouse model. The accumulation of mtDNA alterations in the parkin null background, was shown to increase the serum levels of circulating cell-free mtDNA (ccf mtDNA) and of various cytokines. By contrast, depleting stimulator of interferon genes (STING), which regulates the activation of the DNA inflammasome, sufficed to rescue the degeneration of dopaminergic neurons and a motor impairment previously observed in these animals, suggesting that these phenotypes are the result of inflammatory processes [107]. In a trial experiment as part of this study, we could also show upregulated inflammatory profiles in a small number of PD patients with Parkin mutations [107]. Moreover, Parkin/PINK1 have been shown to modulate cell cycle progression via the downstream target of the cyclic GMP-AMP synthase (cGAS)/STING pathway, TANK-binding kinase 1 (TBK1), at damaged mitochondria. Mitochondrially localized TBK1 is sequestered by Parkin/PINK1 during mitophagy, leading to a block in mitosis. By contrast, loss of Parkin or PINK1 accelerated cellular proliferation in mice [108]. While also NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) has been identified as a target of cGas/STING signaling [109], the inflammasome can equally be activated directly by mitochondrial dysfunction and elevated ROS [110]. Treatment of lipopolysaccharide (LPS)-primed mouse microglia with the mitochondrial complex I inhibitor rotenone induced NLRP3 activation, ASC (apoptosis-associated speck-like protein containing a CARD domain) speck formation and pro-interleukin-1β processing in a concentration-dependent manner [111]. Moreover, enhanced Parkin-mediated ER-mitochondrial tethering and subsequent mitochondrial calcium overload [112] as well as blockage of mitophagy [113] have been reported to trigger NLRP3 inflammasome activation.
In addition to their role in innate immunity, Parkin and PINK1 may also be involved in the control of the adaptive immune response. In mice lacking parkin or pink1, treatment with the bacteria-derived endotoxin LPS [114] or an intestinal infection with gram-negative bacteria [115] induced the formation of MDVs [63], which transport mitochondrial antigens to the plasma membrane, where they are presented on major histocompatibility complex class I (MHC I) molecules [114, 115]. Both processes, MDV induction and mitochondrial antigen presentation (mitAP), are depending on Sorting nexin 9 (Snx9), the cellular abundance of which is regulated by Parkin in a proteasome-dependent manner [114]. Taken together, these findings suggest that Parkin and PINK1 are critically involved in the orchestration of mitophagy induction, immune surveillance and cell cycle control in the context of PD.
CROSSTALK BETWEEN MITOCHONDRIA, LYSOSOMES AND ER AND ITS IMPACT ON CALCIUM HOMEOSTASIS
Multiple lines of evidence suggest that impaired lysosomal degradation causes an accumulation of dysfunctional mitochondria in PD [3]. Mutations in LRRK2 [116] and SNCA [117] have been demonstrated to interfere with lysosomal pathways. Furthermore, in DJ-1-mutant iPSC-derived neurons, mitochondrial stress was shown to trigger oxidized dopamine accumulation, which in turn led to lysosomal dysfunction, and eventually the accumulation of alpha-synuclein [70].
In addition to the crosstalk between lysosomes and mitochondria, the ER is involved in the inter-organellar communication in PD. Alterations of the MAM have been described in different PD models [118]. Exemplarily, alpha-synuclein can be found at the MAM, and pathogenic mutations in SNCA lead to increased mitochondrial fragmentation [119].
Furthermore, calcium homeostasis depends on a well-orchestrated signalling between mitochondria, the lysosome and the ER. In SNCA overexpression models and patient-derived neurons with a triplication mutation, a reduced connection between ER and mitochondria leads to a calcium-dependent decrease in ATP production [120]. However, also Parkin [121], PINK1 and LRRK2 [122], as well as DJ-1 [123] may function in calcium-related pathways.
Emphasizing the role of calcium homeostasis in PD, research demonstrated that isradipine, a calcium channel antagonist, protects dopaminergic neurons [124] by lowering mitochondrial oxidative stress and by reducing mitochondrial turn over and mass [125].
IMPLICATIONS FOR GENETIC TESTING AND POTENTIAL THERAPEUTIC OPTIONS TO AMELIORATE MITOCHONDRIAL FUNCTION IN PD
Currently, only genetic testing allows identifying patients with probable mitochondrial dysfunction by detection of variants in genes associated with mitochondrial pathways. Nevertheless, at present, only a minority of PD patients undergo genetic testing.
A variety of drugs are used in clinical practice to treat PD, mostly by increasing dopamine levels in the midbrain [126]. However, these approaches only allow for symptomatic treatment, and no neuroprotective effect has been demonstrated with any of the drugs approved to date. Such disease-modifying treatment options are urgently needed as neurodegeneration progresses during the disease course, and symptomatic treatment is not able to prevent severe disability and a significant decrease in the quality of life in later disease stages [127].
Various therapeutic approaches focus on a possible mitochondrial etiology of PD: First, several approaches target the presence of ROS. Although positive effects were observed with various substances in vitro and in vivo in animal models, only the antioxidant substance MitoQ that was reported to protect dopaminergic neurons in 6-OHDA-treated mice [128] reached the testing in clinical trials. Unfortunately, there was no evidence for neuroprotection in PD patients [129].
Second, approaches with mitochondrial enhancers, i.e., substances that generally improve the function of mitochondria, were investigated. Particularly noteworthy in this context are studies in which PD patients were treated with coenzyme Q10 in randomized double-blinded trials [130]. However, no effect of coenzyme Q10 administration on neuroprotection was demonstrated in genetically non-stratified patients. Thus, current approaches are based on the assumption that only a subset of PD patients, namely such suffering from a “mitochondrial form of PD”, may benefit from therapy with coenzyme Q10. For this, patients with autosomal recessively inherited PD due to mutations in Parkin and PINK1 could serve as “positive controls”. A current clinical investigator-initiated study based on this principle divides IPD patients using a genomic approach into patients with high and low probability of mitochondrial dysfunction due to the presence of a polygenic risk score composed of mitochondrially associated single nucleotide polymorphisms (SNPs) [131]. Another potential mitochondrial enhancer is vitamin K2. This substance represents, as well as Coenzyme Q10, a dietary supplement. In Drosophila, vitamin K2 has a strong effect on rescuing motor disturbances in pink1 knockout flies [132]. However, studies failed to demonstrate a role for this compound as an electron carrier in mammalian cells [133, 134].
Besides the mentioned established “mitochondrial enhancers”, there are novel compounds that have the potential to ameliorate mitochondrial function in PD patients. For example, a study testing the potential of the neo-substrate kinetin triphosphate (KTP) demonstrated an increase in the kinase activity of mutant PINK1 in cell culture experiments [135], warranting further tests in PINK1 animal models.
Third, selective MAO-B inhibitors like selegiline and rasagiline represent a group of drugs approved for PD treatment, which show possible evidence for a neuroprotective effect. As described earlier, MAO-B is responsible for the processing of MPTP to MPP+, and, therefore, inhibition of this enzyme might reduce oxidative stress. Early after the description of selegiline, findings from animal models suggested a neuroprotective effect [7, 136] and a clinical trial was initiated investigating the effects of selegiline as well as of tocopherol (vitamin E). Here, the so-called DATATOP study suggested a disease-modifying effect of selegiline but not of tocopherol in early stages of PD [137]. However, as selegiline also exhibited symptomatic effects increasing levodopa levels, its neuroprotective effect was questioned. Later, the ADAGIO trial investigated the newer MAO-B inhibitor rasagiline and suggested neuroprotective features in low-dose administration. Surprisingly, this effect was absent at a higher dose [138]. Together, the disease-modifying effect of selective MAO-B inhibitors remains controversial [139]. Furthermore, targeting the interplay between mitochondrial pathways and calcium homeostasis, a clinical trial investigated the calcium channel antagonist isradipine. However, no beneficial effects on motor and non-motor features of PD could be observed [140].
In the context of monogenic PD, the function of the encoded proteins provides a potential starting point for gene-specific therapies [141]. Finally, new treatment options might result from the currently discovered mechanistic relationship between (monogenic) PD and inflammation [107]. In keeping with this notion, the intake of ibuprofen was found to reduce the risk of developing PD [142, 143]. However, further clarification is needed whether inflammation contributes to neurodegeneration in PD, or is instead a consequence of neuronal loss.
CONCLUSION AND OUTLOOK
Mitochondrial dysfunction represents a well-established mechanism in the pathogenesis of both idiopathic as well as monogenic PD. In recent years, investigating monogenic PD has decisively contributed to the clarification of impaired mitochondrial pathways in the sporadic disease. In light of the manifold literature on this topic, it is tempting to speculate that several of the above-mentioned PD proteins form a pathophysiological network surrounding mitochondria. Alterations at any point of this network may contribute to the disease, although the exact mechanisms orchestrating this interplay are still not fully understood.
Despite our advances in basic PD research, clinical trials targeting mitochondrial dysfunction and oxidative stress have not demonstrated significant beneficial effects to date. Of note, however, patients have not yet been stratified according to the etiology of disease in previous trials. In the meantime, different etiologic subtypes of PD have emerged. Stratification approaches, according to such specific subtypes of the disease, are currently being developed and incorporated into trial designs [131].
Most recently, a link between immunologic alterations and mitochondrial dysfunction in autosomal recessively inherited monogenic PD has been demonstrated [107]. However, evidence that inflammation causes neurodegeneration is limited thus far, and the role of immunity in PD needs further elucidation. Regarding monogenic PD in general, first gene-specific therapies allowing personalized treatment are already undergoing clinical trials. Together, further in-depth investigation along with biomarker establishment of a “mitochondrial subtype” of PD represents a promising approach to arrive at a more individualized treatment even of IPD patients. In the future, continuous efforts in both basic and clinical research with a fast translation of new insights into clinical practice have the potential to lead to new therapeutic approaches in “mitochondrial PD”.
CONFLICT OF INTEREST
CK serves as medical advisor for genetic testing reports in the fields of movement disorders and dementia, excluding Parkinson’s disease, for Centogene. MB, SLR and AG have no competing interests to declare.
Footnotes
ACKNOWLEDGMENTS
CK is supported by SysMedPD (European Union’s Horizon 2020 research and innovation program under grant agreement 668738). CK and AG are supported by the German Research Foundation (FOR2488, GR 3731/5-1). AG received funding from the Luxembourg National Research Fund (ATTRACT career development grant, FNR9631103; INTER grant, FNR11250962).