
Abstract
Parkinson’s disease (PD) is a slowly progressing neurodegenerative disorder that is coupled to both widespread protein aggregation and to loss of substantia nigra dopamine (DA) neurons, resulting in a wide variety of motor and non-motor signs and symptoms. Recent findings suggest that the PD process is triggered several years before there is sufficient degeneration of DA neurons to cause onset of overt motor symptoms. According to this concept, the number of DA neurons present in the substantia nigra at birth could influence the time from the molecular triggering event until the clinical diagnosis with lower number of neurons at birth increasing the risk to develop the disease. Conversely, the risk for diagnosis would be reduced if the number of DA neurons is high at birth. In this commentary, we discuss the genetic and epigenetic factors that might influence the number of nigral DA neurons that each individual is born with and how these may be linked to PD risk.
Keywords
INTRODUCTION
Parkinson’s disease (PD) is a progressive neurodegenerative disease that is associated with a characteristic set of motor and non-motor disturbances. Most cardinal motor symptoms such as bradykinesia, rigidity, and postural instability, and to a lesser extent tremor, are considered largely a consequence of loss of striatal dopamine (DA), secondary to the degeneration of DA neurons in the substantia nigra [1]. Another major neuropathological finding is widespread accumulation of alpha-synuclein (α-syn) in neuronal perikarya and neurites [2], which is believed to contribute to both motor and non-motor deficits. These disease features are apparent in common idiopathic PD, as well as rare familial cases with single point mutations or gene duplications and triplications of α-syn [3]. Several other autosomal dominant (with variable penetrance) and recessive familial PD genes have been identified, although a definitive disease mechanism has not yet been identified for these mutations [4]. In addition, a growing number of single nucleotide polymorphisms are known to influence PD risk [5]. These genetic loci clearly influence disease risk in the approximate 90% of PD patients that are classified as having idiopathic disease. Notably, while heritability has been estimated to underlie around 25% of PD risk [6], environmental factors and age are more impactful on disease risk in sporadic cases [7]. It is believed that the loss of striatal DA, with concomitant degeneration of nigral DA neurons, has to exceed a certain threshold before motor symptoms are evident, and the clinical diagnosis of PD can be made. Therefore, the number of nigral DA neurons that are present at birth might influence the lifetime risk of being diagnosed with PD. The purpose of this short review is threefold. First, to discuss the literature describing variability in numbers of nigral DA neurons between normal individuals. Second, to consider genetic as well as epigenetic/environmental factors that can influence this variability. Third, to propose a model for how the variability can impact lifetime PD risk.
INTER-INDIVIDUAL DIFFERENCES IN DA CELL NUMBERS
In this section, we first describe estimates of the proportion of nigral DA neurons reported to have died in PD patients coming to autopsy, and then we discuss the variation in number of nigral DA neurons found in brains of normal subjects.
The average reduction of nigral DA neurons determined by stereological estimates in 181 PD patients across 12 studies has been estimated to around ∼68% but reflected considerable inter-study and inter-individual variation [8]. The focus has not been on the absolute number of nigral DA neurons remaining in the PD patients’ brains, but instead it has been on the number of remaining neurons expressed as a percentage of the numbers found in normal healthy subjects from the same study. Generally, it is believed that the absolute number of functional DA neurons remaining in the substantia nigra, not yet affected by the PD process, that decides when the “tipping point” is reached and significant clinical motor symptoms appear. The degeneration of nigral DA neurons considered to be progressive, in a linear or stepwise fashion, and starts many years before the first motor symptoms. Because there are no datasets of nigral DA neuron counts available from individuals who had recently exhibited onset of PD symptoms, it is not possible to state with confidence how many neurons must die before motor symptoms appear. These assumptions imply that the number of DA neurons that an individual is born with could influence the lifetime risk for PD.
Considering the potential importance of starting number of DA neurons for PD risk, it is pertinent to ask how many DA neurons are present in the normal human substantia nigra? Surprisingly, there is not a strong consensus on this in the literature. Early stereological studies of substantia nigra neurons in normal healthy humans have generally quantified neuromelanin-containing neurons (pigmented) on Nissl-stained tissue. This number generally correlates well with the number of tyrosine hydroxylase (TH) neurons [9], though age-related changes including the buildup of intracellular neuromelanin [10], and increases in monomeric α-syn [11] may lead to phenotypic down-regulation in viable nigral neurons leading to discrepancies between the number of pigmented and TH-immunoreactive neurons in the same individual. The longstanding idea that the number of midbrain DA neurons present at birth might affect susceptibility to PD [12] was initially based on observations of mouse strain differences in the number of nigral TH-immunopositive neurons [12–14]. We assessed the literature to define a natural variation in the number of DA neurons in the substantia nigra of healthy humans. We focused on studies employing stereological approaches to quantify the numbers of pigmented neurons in the substantia nigra and observed a considerable variation in human subjects across four studies [9, 15–17] (Fig. 1A). To avoid confounding effects in the healthy human controls, the authors of the studies followed strict exclusion criteria such as history of neuropsychiatric diseases and/or presence of neuropathology with only minor differences. In the data presented by Pakkenberg et al. [15] the difference between the healthy human subjects with the lowest and highest number of pigmented neurons reached 152% (∼4.00–6.10×105 neurons), while the study by Ma et al. [16] revealed 433% in difference between the highest and lowest population size (∼0.75–3.25×105 neurons). Cabello et al. [9] and Rudow et al. [17] presented ranges of 372% (∼1.74–6.49×105 neurons) and 220% (∼2.32–5.13×105 neurons), respectively. We also specifically focused on data from individuals who died during the first five decades (18–50 years) to minimize the risk that any variance in cell number was due to aging or early stages of age-related, progressive disorders not yet discernable (Fig. 1B) [18]. In the 18–50 years dataset, the variation in the number of pigmented neurons in the nigra was still high. Across the studies listed in Fig. 1B, Ma et al. [16] reported a 185% difference in the subjects with lowest and highest number of pigmented neurons in the substantia nigra (∼1.75–3.25×105). Cabello et al. [9] reported a 293% difference (∼2.21–6.49×105) and Rudow et al. [17] reported a 147% difference (∼3.48–5.13×105) in the number of pigmented neurons (Fig. 1B). In short, all the available data sets we examined showed considerable natural variation in the numbers of nigral DA neurons.
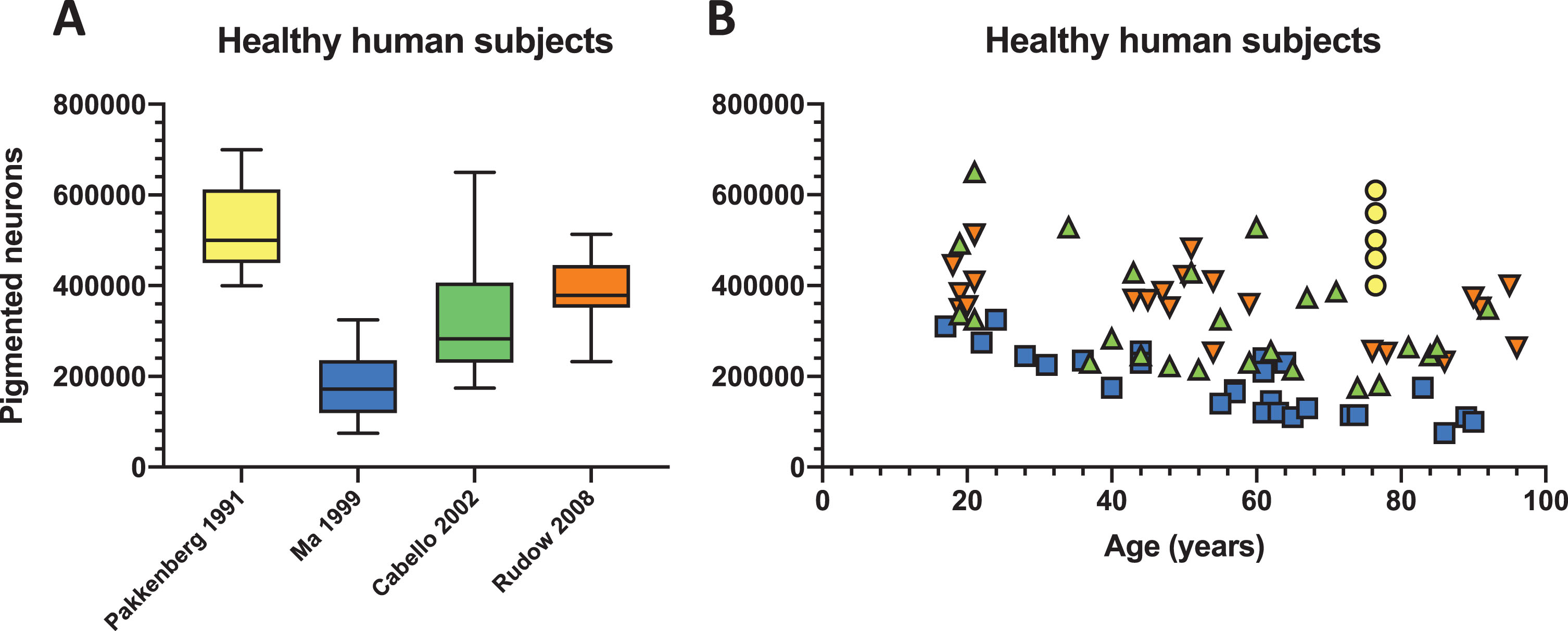
Natural variation in the number of pigmented neurons in healthy human. Four studies employing stereological quantification of the number of nigral pigmented neurons [9, 15–17] were selected and (A) depicted in a box and whisker plot showing considerable variation in healthy human subjects. (B) The same data were also plotted against ageing where the first five decades where the brain should be relatively unaffected by various confounders (e.g., ageing) retains a high degree of variability in the number of pigmented neurons. Some aged individuals in the eighth and ninth decade show a very high number of pigmented neurons comparable to individuals in their twenties.
DOES THE INHERITED NUMBER OF NIGRAL DOPAMINE NEURONS AFFECT PD RISK?
As mentioned above, the threshold for PD motor symptoms might be influenced by the starting number of nigral DA neurons at birth, or the number of cells that survive the immediate post-natal pruning of the nigrostriatal system (Fig. 2). In this section, we discuss this concept in more detail. The idea of high variability among DA neurons and its clinical implications was conceived more than five decades ago. It was based on observations showing considerable differences among inbred mouse strains relating to TH activity [19], DA neuron numbers and striatum size [12, 20]. Recently, the genetic and epigenetic pathways that govern the development of nigral DA neurons have shed more light on this research area. Formation of midbrain DA neurons is tightly orchestrated by the tempo-spatial expression of a group of highly conserved transcription factors (e.g., EN1/2, OTX2, GLI1/2, FOXA1/2, LMX1A/B, MSX1/2, NEUROG1, ASCL1 and NATO3) and morphogens (e.g., FGF8, WNT1 and SHH) that shape the rostro-caudal and dorso-ventral identities [21, 22]. Many of these genes are expressed in fully mature DA neurons, together with NURR1 and PITX3, and are involved in adult neuron maintenance. Especially NURR1 is prerequisite for the expression of TH and SLC6A3 that defines the midbrain DA phenotype [23]. Consequently, the substantia nigra DA population size is not only defined by a predetermined gene program, but also through how efficient cellular maintenance is for evading potential stress-related cell loss. This may further explain why polymorphisms in NURR1 and PITX3 have been linked with PD risk [24, 25]. Insufficiency in transcription factor genes that govern nigral DA neuron development has been associated with rare developmental abnormalities in humans [26] and further detailed investigations in vivo specifically implicate these genes in determining the anatomical location, formation and size of the DA neuron population [22]. Ectopic or increased expression of genes such as FOXA2, LMX1A [27, 28], OTX2 [29] and CTNNB1 [30] change the anatomical location or increase the size of the midbrain DA population in vivo. Mutations in the primary sequence of these genes are unlikely to be the source of the size variation in the midbrain DA population. Instead, fine-tuning of gene transcription via cis-regulating elements [31] is a possible central determinant of the inter-individual variation in the number of the substantia nigra neurons. Considering that the epigenetic landscape changes considerably from progenitor into post-mitotic neuron [32], it will be challenging to link the activation state, mutations or single nucleotide polymorphisms in these non-coding regions during embryogenesis with the number of DA neurons that are present at birth. Some tentative clues are appearing in the literature. In cells derived from human embryonic stem cells (hESC), a study recently tracked PD risk single nucleotide polymorphisms to the disruption of enhancers important for transcriptions factors involved in mesodermal differentiation [33]. One particular important mesodermal structure is the notochord that releases Shh, which is a secreted signaling molecule essential for the induction of the floor plate and hence the development of midbrain DA neurons. [33]. This suggests a potential link between a PD risk single nucleotide polymorphism and epigenetic regulation of genes involved in the determination of the number of substantia nigra DA neurons. Studies characterizing induced pluripotent stem cells (iPSCs) have offered a potential link between a low number of nigral DA neurons and a PD mutation. Neural stem cells (NSCs) differentiated from iPSCs generated from an early onset PD patient with a PLAG26 mutation showed profound reduction in proliferation and differentiation of DA neurons in vitro [34]. Similar observations of reduced proliferation and differentiation capacity were observed in NSCs derived from LRRK2 mutant (G2019S) iPSCs after prolonged passaging [35]. Studies of DA neurons differentiated from iPSCs derived from sporadic and familial PD (LRRK2 G2019S, PINK Q456X and triSNCA) have demonstrated that the deficiencies observed in PD-derived NSCs seems to be passed on to the progeny [34, 36–39]. These changes were only evident upon differentiation into DA neurons after prolonged culturing [40]. The methylation profiles of the cells derived from iPSCs from familial PD cases resembled cultures not enriched in DA neurons, suggesting an inherent inability to fully adapt the epigenetic identify of a healthy DA neurons [40]. These findings were integrated in a theoretical model where PD-related enhancer methylation was associated with the downregulation of a transcription factor network involved in neurogenesis and survival (HNF4A, FOXA1, NR3C1 and FOSL2) and upregulation of a transcription factor network (OTX2, PAX6 and ZIC1) and genes (SNCA, DCC and DCT) involved in proliferation, differentiation and survival (via PAX6) [40]. Cell death is a naturally occurring event in the formation of the nigrostriatal circuitry at around post-natal day 2 and 14 in rodent nigral DA neurons [41]. Therefore, a reduced ability of PD iPSCs to tolerate stress in vitro might reflect how well the DA neurons are equipped to survive during early development in vivo, either during embryogenesis or shortly after birth.
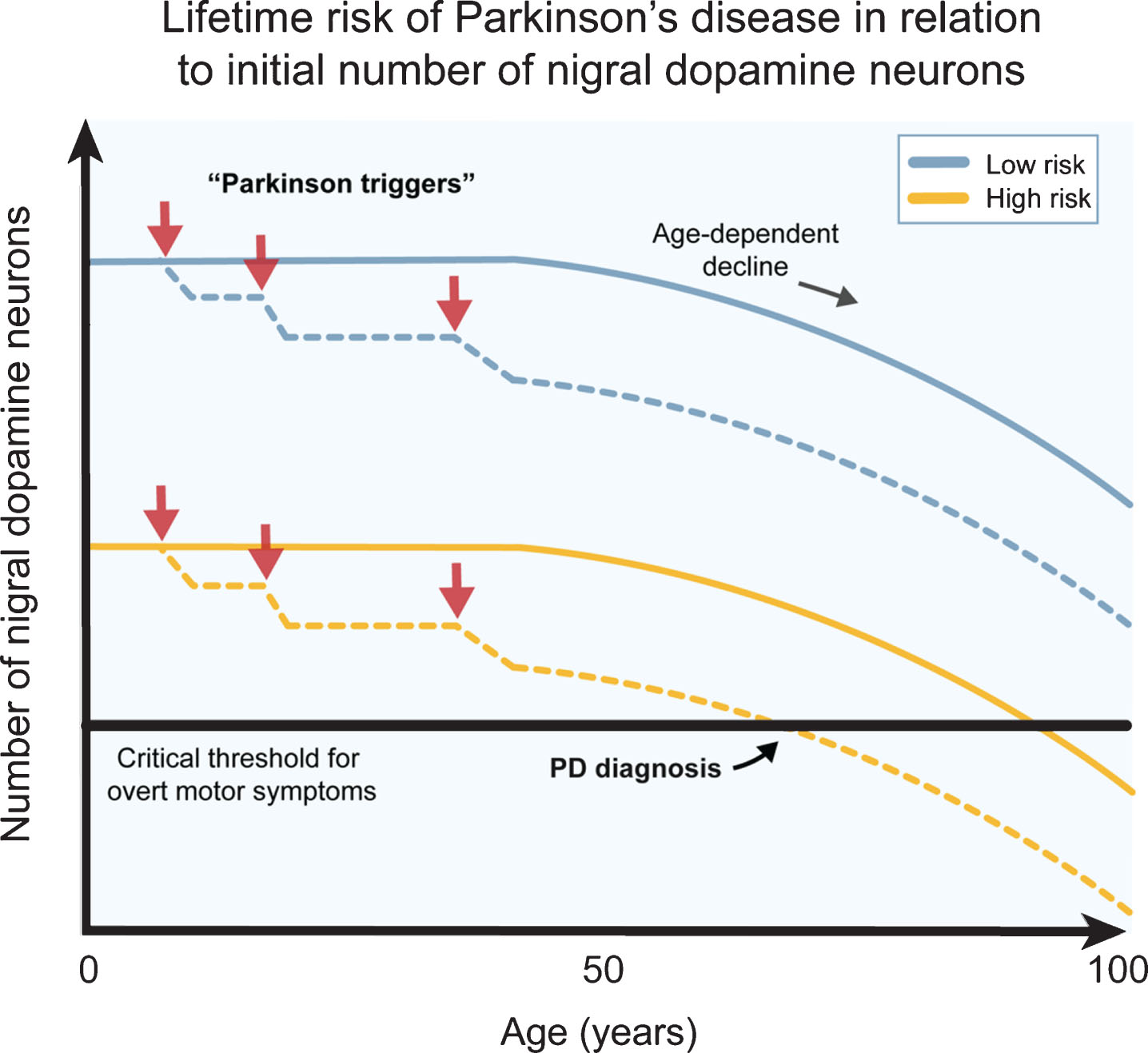
The number of dopamine neurons and Parkinson’s disease risk. Several genetic, epigenetic and non-genetic factors affect the generation of dopamine (DA) neurons and their survival during development and birth. This likely contributes to a natural high variation in healthy human subjects. Individuals born with higher number of DA neurons are more robust to Parkinson’s disease (PD) triggers (red arrows) since they can afford a higher cell loss before onset of motor symptoms (blue lines), while individuals born with a smaller starting population of DA neurons can afford a much lower cell loss from the exposure to PD triggers before the onset of motor symptoms (yellow lines). The combination of PD triggers and the natural age-related decline of DA neurons may therefore put some individuals at a greater lifetime risk of acquiring PD motor symptom. The DA neuron pool from birth may therefore be an important parameter when considering PD risk.
A role of α-syn in PD pathogenesis is well documented in the adult brain, but less so in early development. An in vivo study showed that the number of TH-immunoreactive neurons in the substantia nigra was affected by the expression of α-syn in a gene dose dependent manner in mice [42]. High expression of α-syn led to increased numbers of nigral TH-immunoreactive neurons in nigra and vice versa [42]. Interestingly, the effect of removing α-syn expression specifically caused a reduction in the number of TH-immunoreactive neurons at embryonic day 13.5 (but not at day 10), approximately coinciding with the ontogenetic pruning of DA neurons that has been described to occur at embryonic day14 [42]. This may be related to the reported ability of α-syn to increase tolerance to oxidative stress [43, 44], and may consequently exert an important function during development and survival of DA neurons.
THE ROLE OF NON-GENETIC FACTORS IN AFFECTING THE DA POPULATION BEFORE AND AT BIRTH
Non-genetic factors in utero can also impact the critical periods of brain development when the DA neuron population is born and undergoes maturation, as well as the time window when a subset of the DA neurons is selected for developmental programmed cell death. Prenatal infections with influenza virus have been associated with increased risk of neuropsychological diseases and PD and is paralleled directly with apoptosis of DA neurons in the nigra [45]. Maternal inoculation with lipopolysaccharide is detrimental to TH neurons around E10.5 in the rat fetus [46] which is around the time that TH expression is turned on in the floor plate (E10.5–12.5) [22]. Exposure to environmental toxins during pregnancy or hypoxic conditions at birth may similarly affect DA neurons by disturbing mitochondrial function which is essential for proper neurogenesis and differentiation [47]. In addition to changing the number of DA neurons surviving at birth, or through the developmental period shortly thereafter, these factors might further impact sensitivity to additional insults that could occur in adulthood [48].
CONCLUDING REMARKS
The DA neuron is undoubtably in the front line when it comes to understanding PD risk genes, epigenetic changes and environmental factors (Fig. 2). In this commentary, we highlight that some of the genetic loci that now are known to influence PD risk might not impact death processes in dopamine neurons in the adult organism. Instead, we propose they influence lifetime risk of developing motor symptoms by affecting the number of nigral DA neurons that each individual is born with or that survives immediate postnatal development. The cell number that each individual has when leaving infanthood might further depend on non-genetic and non-epigenetic factors such as maternal infections and endogenous or environmental toxins that impact intrauterine health. Our most important take home message is that we need to explore changes that occur both during development and in during adulthood and aging when we seek to understand the full landscape of PD risk.
CONFLICT OF INTEREST
P.B. has received commercial support as a consultant from Axial Biotherapeutics, CuraSen, Fujifilm-Cellular Dynamics International, IOS Press Partners, LifeSci Capital LLC, Lundbeck A/S and Living Cell Technologies LTD. He has received commercial support for grants/research from Lundbeck A/S and Roche. He has ownership interests in Acousort AB and Axial Biotherapeutics and is on the steering committee of the NILO-PD trial. J.H.K has received commercial support as a consultant from Cellular Dynamics International, Inc, Michael J. Fox Foundation, Abbvie, Exicure, NSGENE, Guidepoint, Inhibikhase, Axovant, and Seelos.
Footnotes
ACKNOWLEDGMENTS
P.B. is supported by grants from the National Institutes of Health (1R01DC016519-01, 5R21NS 093993-02, 1R21NS106078-01A1). P.B. reports additional awards from Office of the Assistant Secretary of Defense for Health Affairs (Parkinson’s Research Program, Award No. W81XWH-17-1-0534), and the Peter C. and Emajean Cook Foundation, which are outside but relevant to the submitted work.