
Abstract
Parkinson’s disease (PD) is a neurodegenerative disorder with complex etiology and variable pathology. While a subset of cases is associated with single-gene mutations, the majority originates from a combination of factors we do not fully understand. Thus, understanding the underlying causes of PD is indispensable for the development of novel therapeutics. Glycation, the non-enzymatic reaction between reactive dicarbonyls and amino groups, gives rise to a variety of different reaction products known as advanced glycation end products (AGEs). AGEs accumulate over a proteins life-time, and increased levels of glycation reaction products play a role in diabetic complications. It is now also becoming evident that PD patients also display perturbed sugar metabolism and protein glycation, including that of alpha-synuclein, a key player in PD. Here, we hypothesize that anti-diabetic drugs targeting the levels of glycation precursors, or promoting the clearance of glycated proteins may also prove beneficial for PD patients.
PARKINSON’S DISEASE IS A MULTI-FACTORIAL DISEASE WITH UNCLEAR ETIOLOGY
Parkinson’s disease (PD), the second most common neurodegenerative disease, is typically known for the loss of dopaminergic neurons in the substantia nigra pars compacta and for the occurrence of Lewy bodies and Lewy neurites (intracellular proteinaceous inclusions) in the surviving cells [1]. Since its initial description by James Parkinson in 1817, it is now widely accepted that PD is not simply a movement disorder. Apart from the defining motor features—rigidity, resting tremor, bradykinesia, and disturbed gait—the majority of PD patients also experiences one or several non-motor features that may precede the onset of motor symptoms by decades. Non-motor features include olfactory dysfunction, constipation and other disturbances of the digestive system, REM behaviour disorders, dementia, cognitive decline, anxiety and depression [2]. The observation that Lewy-body pathology and other pathological changes can be detected in the gut and/or in the olfactory system many years before motor-features, led to the hypothesis that the PD-pathology spreads from the peripheral nervous system to the CNS [3].
PD is an extremely heterogeneous disorder, with different ages of onset, progression rate, pathology and disease manifestation. Thus, several disease subtypes are pooled together as PD [4]. In addition, several related disorders including multiple systems atrophy and dementia with Lewy bodies share common features with PD. The classification of PD is often based on motor features, but non-motor features and age of onset are also used to sub-classify PD cases [2]. However, a definitive detailed categorization of PD cases is still not consensual. Attempts to classify PD cases based on more objective tests such as biomarkers or neuroimaging are expected to lead to major advances in the field. Subdividing PD into sporadic and monogenic forms has already led to the identification of groups with more similar clinical features. Extending this effort to ensure improved discrimination between subtypes of PD will also prove critical both for fitting therapeutic approaches to individual patients, and for discovering the molecular underpinnings of PD pathology, which may in turn constitute novel targets for therapeutic intervention.
Most PD cases are sporadic, and only 5–10% have a known genetic cause and can be ascribed to mutations in single genes [1]. The first gene associated with monogenic PD was SNCA, encoding for the protein alpha-synuclein (aSyn), the main component of Lewy bodies and Lewy neurites, thought to play a central role in the disease [5, 6].
A number of genetic and environmental risk factors for PD/parkinsonism have been identified over the years. For example, the exposure to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) was found to cause a PD-like disease [7]. Other environmental risk factors, such as rural living, well water consumption, exposure to pesticides or solvents, were also associated with some forms of parkinsonism [8–10]. On the other hand, coffee or nicotine consumption are associated with a decreased risk to develop PD [11, 12]. Given that the majority of PD cases are caused by a combination of both genetic and environmental factors, it is critical to analyse in detail the impact of these environmental factors on the molecular mechanisms that lead to PD pathology. In this context, glycation emerges as a possible environmental factor that can impact on PD pathogenesis.
GLYCATION OF PROTEINS: AN UNAVOIDABLE PROCESS
Glycation, via the Maillard reaction, named after Louis C. Maillard who first described the browning processes in food processing [13], involves the non-enzymatic reaction of reducing carbohydrates and amino compounds. In fact, it is not a single reaction but a complex series of reactions that was studied for decades, mainly in the context of food browning and is lately also receiving attention due to its role in neurodegenerative diseases [14, 15]. These Maillard reactions were characterized and grouped into a scheme [16]. In the first step, a carbonyl group (from glucose or other reactive carbonyl species) condenses with an amino or thiol group of amino acids, nucleic acids or amino lipids, giving rise to an early glycation product termed Amadori compound. In the second step of the reaction, the intermediate Amadori compound is rearranged and breaks down in one of several possible chemical pathways. A variety of different carbonyl and dicarbonyl intermediate products, including glyoxal and methylglyoxal (MGO) are formed, and can exist free or bound to proteins. Lastly, higher molecular weight species (advanced glycation end products (AGEs)) are formed from these lower molecular weight intermediates [17]. Importantly, other pathways and mechanisms including oxidative stress also contribute to the formation of reactive carbonyl species [18]. As mentioned above, glycation can affect various biomolecules. While proteins are important targets of glycation (the N-terminal residue, the amino group of lysine residues, and the guanidinium group of arginine residues can be subject to Maillard modifications), DNA [19, 20] and lipids [21] can also be subject to glycation.
The term “glycation” is often used in a rather imprecise manner. It is frequently used to distinguish the non-enzymatic reaction from the enzymatic glycosylation of proteins that go through the secretory pathway. Furthermore, glycation may refer specifically to the formation of an Amadori product, via the reaction of an amino acid with glucose. But the term glycation is also used to describe the addition of carbonyls, such as MGO, to proteins and the formation of AGEs. This inconsistent nomenclature reflects the lack of in-depth understanding of the mechanisms and physiological roles of these reactions.
The most abundant AGEs are MGO-derived hydroimidazolone 1, the lysine arginine cross-link glucosepane and N(ɛ)-carboxymethyllysine (CML) [22]. However, it is critical to appreciate that the Maillard reaction network can give rise to hundreds or thousands of different products. The reaction of only one sugar with one amino acid was recently shown to lead to the formation of up to 300 products [23], and this immense variety of adducts with different characteristics complicates the study of Maillard reactions. A variety of methods have been applied to study the non-enzymatic reaction of reducing carbonyls and amino compounds. Given that a number of AGEs are fluorescent, it is possible to follow their formation simply by measuring their fluorescence. Furthermore, the availability of antibodies directed to different AGEs enables the analysis of the localization of AGEs in cells and tissues using immunochemical methods [24]. In addition, mass spectrometry techniques are crucial for assessing glycation products, especially since advances in untargeted mass spectrometry enable the detection of a larger number of molecules [25, 26].
The levels of AGEs in a given tissue are determined by the formation rate, the stability of the resulting AGE, and the turnover of the target protein. Even though there are differences between the various routes of the Maillard reaction, the reaction is generally rather slow, and the reaction products are very stable [17]. The degree to which an individual protein undergoes Maillard modifications depends on the amount of precursors and the availability of potential target sites for the modification [22]. While long lived proteins have the chance to accumulate many AGEs, degradation of short lived proteins releases free, non-protein bound AGEs [27].
The majority of dicarbonyls are catabolized by the glyoxalate system, comprising the highly abundant glyoxalases Glo1 and Glo2. MGO is converted to D-lactate in a glutathione-dependent reaction [28]. Other systems that protect cells from carbonyl stress involve aldo-ketoreductases, aldehyde-dehydrogenase and fructosamin-3-kinases [29, 30]. Interestingly, DJ-1, the product of the PARK7 gene that is associated with recessive forms of PD, was recently shown to be associated with glycation. DJ-1 displays various cellular roles, including chaperone function, response to oxidative stress, and glyoxalase activity [31–33]. In addition, it was suggested that DJ-1 can deglycate MGO- and glyoxal-modified amino acids, nucleotides, and nucleic acids [19, 34]. However, these studies remain controversial [35], and the physiological role of DJ-1 as a deglycase remains to be confirmed.
DIABETES AS A RISK FACTOR FOR PD
Diabetes has been suggested as a risk factor for PD and other neurodegenerative disorders. Recent studies suggest that diabetes increases the risk for PD by 23% [36]. After adjusted hazard ratio, a larger magnitude was observed in females, and only diabetic patients over 65 years-old showed increased risk for PD [36]. In this study, diabetes duration was not considered. However, in the Neurological Disorders in Central Spain (NEDICES) study, which included 4919 controls (828 with diabetes) and 79 PD patients (14 with diabetes), the duration of diabetes emerged as an important factor in the association between PD and diabetes. The increased risk might be limited to those with longer disease duration (>10 years) [37]. This was also previously observed in a cohort in the U.S.A. [38]. Nevertheless, it is now established that diabetes prior to PD increases the risk for more severe PD features [39].
Several meta-analysis studies have evaluated diabetes as a risk factor for PD. For example, a recent association study, including a total of 7 population-based cohort studies, further suggests that diabetes is associated with a 38% increase in PD risk [40]. This meta-analysis is consistent with previous studies [38, 42]. Intriguingly, some reports found no association between diabetes and PD [43–47].
Likewise, PD diagnostic, severity, progression and medication should also be considered.
In a recent genome-wide association study (GWAS) to assess the association between immune-mediated diseases and PD, 4 genetic loci were found to be shared between PD and type 1 diabetes [48].
Further evidence linking diabetes with PD comes from prospective studies where diabetes is found to contribute to the development of PD. For example, one of the non-motor features associated with PD is mild cognitive impairment (MCI) [49]. PD patients with diabetes display reduced cortical grey matter, amygdala, frontal white matter and temporal white matter volumes, and higher total white matter hyperintensity and periventricular hyperintensities. This occurs regardless of the duration of PD [50, 51]. In a longitudinal study, PD patients with diabetes presented a higher rate of atrophy in the cortical white matter, particularly in the parietal and occipital white matter. These patients also presented a greater decline in Mini-Mental State Examination (MMSE) and Montreal Cognitive Assessment (MoCA) scores, tests aimed to screen for dementia or MCI [50, 52].
ALTERED HOMEOSTASIS OF SUGAR METABOLISM IS A COMMON FEATURE BETWEEN DIABETES AND PD
The deleterious effects of high neuronal glucose in diabetic conditions are well known. While the brain weighs approximately 2% of the whole body, it accounts for about 20% of the whole energy consumption. Neurons have a constantly high demand for glucose as the preferred energy source. However, neurons can only store minimal amounts of glucose intracellularly [53]. The glucose transporters GLUT1 and GLUT3, that are mainly expressed in the blood brain barrier and in neurons, respectively, efficiently take up glucose from the blood into brain and neurons. Importantly, their function is not modulated by insulin. While certain brain areas also express the insulin dependent GLUT4, it appears that glucose uptake in the brain is insulin-independent [54]. Accordingly, several reports confirmed that intracerebral glucose levels depend solely on those in the plasma in rat and humans [55–57] and it is estimated that the glucose levels in neurons and in the extracellular space are identical [56]. Thus, neurons appear to be particularly exposed to fluctuating glucose levels.
Intracellular glucose is normally phosphorylated to glucose-6-phosphate to enter glycolysis or the pentose phosphate pathway. However, high glucose levels lead to diverging metabolic routes giving rise to reactive dicarbonyl species [58]. Despite advances in the adjustment of blood sugar levels, diabetes patients frequently experience persisting hyperglycemia, or reoccurring episodes [58]. Accordingly, the levels of reactive dicarbonyls and AGEs were shown to be elevated in diabetes patients and in experimental models of diabetes [59, 60]. Even though the overall dicarbonyl concentration in tissue is not high, dicarbonyls are significantly more reactive than glucose [17, 61]. Interestingly, glycation of proteins is thought to be a main cause of several diabetic complications including retinopathy, neuropathy, and nephropathy, and the AGE content in tissues enables predictions about the severity and the occurrence of diabetic complications [62].
Both glucose and MGO levels are elevated in diabetes. However, their ratios are still a subject of discussion, and it is suggested that, in addition to hyperglycemia, other metabolic changes contribute to elevated MGO concentrations [63]. Indeed, Glo1 levels were shown to be decreased in several PD models and in PD patients [64]. Furthermore, Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), a glycolytic enzyme that constitutes 5–20% of total soluble cytoplasmic protein, is subject to glycation, leading to its inactivation. In turn, GAPDH inactivation and altered glycolysis levels lead to redirection of the glucose flux towards the pentosephosphate-pathway, possibly also increasing the rate of dicarbonyl formation [65].
It was also suggested that insulin itself may also bind MGO [58]. Altogether, these findings suggest a critical role of MGO in the global impairment of energy metabolism in diabetes. While hyperglycemia can, in many cases, be corrected, glycation of tissue proteins accumulates over their lifetimes, thereby contributing to the chronic complications that many diabetes patients experience (Fig. 1).
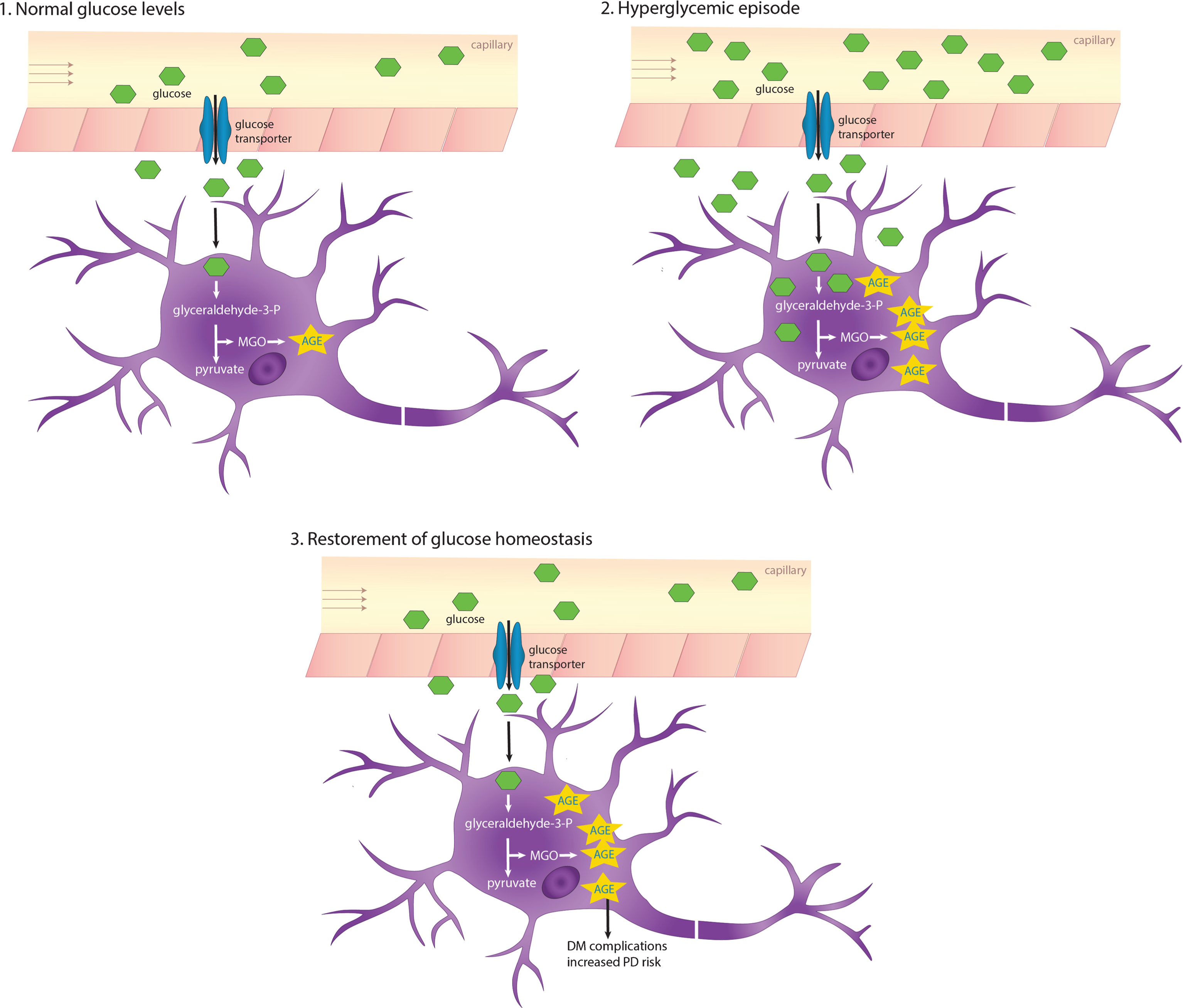
AGEs accumulate due to recurring or chronic hyperglycemia. The glucose transporters Glut1 and Glut3 that are expressed in the blood-brain barrier and neurons, respectively, shuttle glucose in an insulin-independent manner. In hyperglycemic conditions, intracerebral glucose levels also increase, which in turn increases glycation and AGEs accumulating especially in long-lived proteins. DM, diabetes mellitus.
Several studies reported a dysregulation of sugar metabolism in PD. Glucose-6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase, key enzymes in the pentose-phosphate pathway, are decreased in post-mortem PD brains [66]. PD patients display decreased glucose tolerance and hyperglycemia. Furthermore, elevated levels of the monosaccharides fructose and mannose are present in the cerebrospinal fluid of early-stage PD patients [67, 68]. Along this line, aSyn deficient mice display altered AGEs and increased Glo1 activity, further illustrating aSyn involvement in sugar metabolism [69].
Several human diseases, including PD, are associated with protein misfolding and aggregation [70]. aSyn, a key player in PD, is a long-lived protein and, therefore, is likely to be glycated over time [71]. In fact, AGEs colocalize with aSyn in Lewy bodies in the substantia nigra [72, 73], and we have recently identified glycated aSyn in brain tissue from PD patients [74]. Furthermore, it was shown that aSyn can be glycated in vitro, in the N-terminal region, by dicarbonyl compounds and ribose [74–76].
The physiological function of aSyn is affected by glycation in several ways: It was shown both in human cell lines and in animal models that glycated aSyn is more prone to oligomerization (Fig. 2) [74, 77]. Furthermore, studies in a PD cell models revealed that glycated aSyn displays reduced mono-ubiquitination, proteasome- and autophagy-lysosome-mediated degradation [74]. In addition, MGO-treatment was shown to lead to reduced aSyn release and impaired binding to lipid membranes, further contributing to accumulation of aSyn inside cells [74]. Interestingly, aSyn physiology is not just altered directly by glycating agents. Actually, the presence of AGEs produced, for example, from bovine serum albumin, induces the cross-linking of aSyn [78]. Despite progress made on unrevealing the complex effects of glycating agents on aSyn physiology and pathology, a number of questions remain.
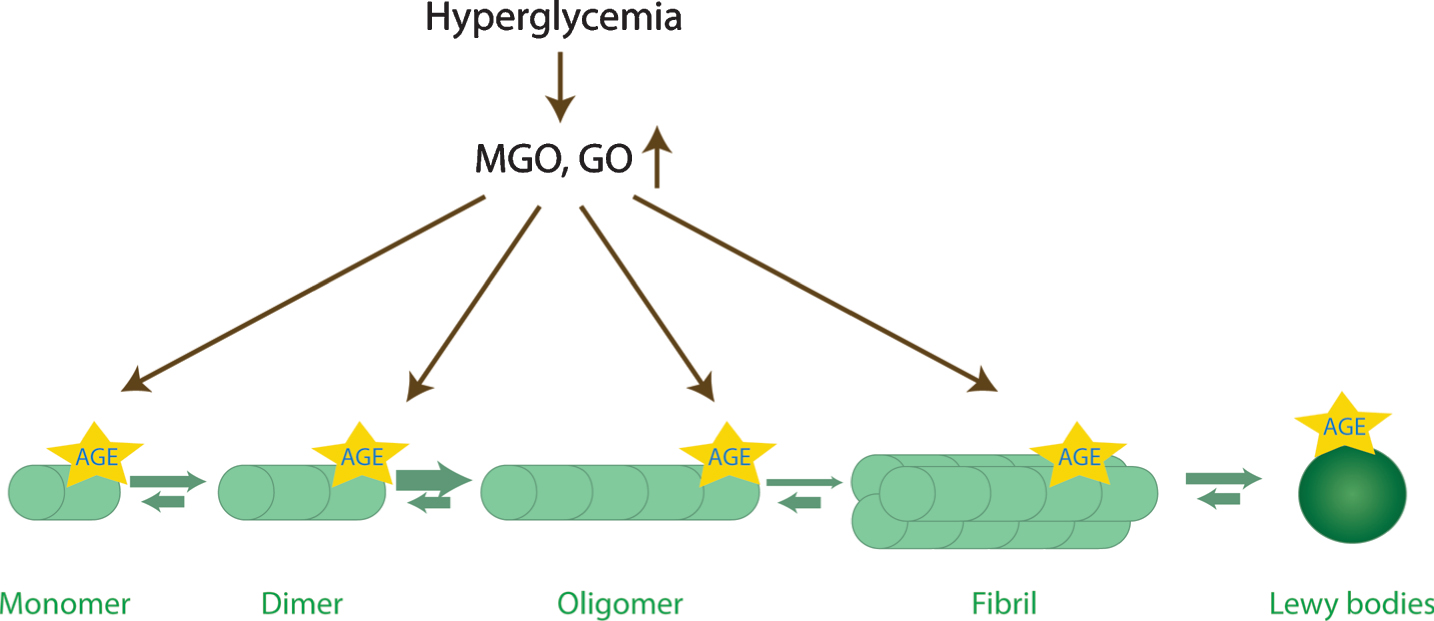
Glycation affects aSyn conformations. aSyn is the predominant component of Lewy bodies. During the aggregation process, aSyn monomers start to assemble into dimers, oligomers, and eventually into β-sheet containing fibrils. Oligomers are currently thought to be the most toxic species. Interestingly, the levels of reactive dicarbonyls (GO, glyoxal; MGO, methylglyoxal) increase upon hyperglycemia, and may potentiate aSyn oligomerization, while inhibiting its fibrillization, and lead to the accumulation of toxic glycated aSyn oligomers.
DOPAMINE AND MGO: PARTNERS IN CRIME
The loss of dopaminergic cells in the substantia nigra pars compacta is a hallmark of PD, but it is unclear why this particular type of dopaminergic neurons appear particularly more vulnerable. The “auto-toxicity theory” suggests that dopamine products itself are responsible for the degeneration of dopaminergic neurons. The catecholamine dopamine can undergo several chemical transformations that give rise to toxic molecules. In particular, spontaneous oxidation leads to the formation of reactive oxygen species and quinones. Enzymatic oxidation of dopamine catalysed by monoamine oxidase gives rise to a highly toxic and reactive intermediate compound: 3,4-dihydrox-yphenylacetaldehyde (DOPAL) [79]. Dopamine-derived quinones were shown to react with several proteins implicated in PD, including aSyn, parkin, DJ-1 [80–82]. Interestingly, it was shown that DOPAL leads to the oligomerization of aSyn which alters synaptic vesicle homeostasis [83]. DOPAL can be metabolized enzymatically by aldehyde dehydrogenase. Dopamine-derived metabolites share reactive groups with MGO: dicarbonyl groups drive the reaction with amino groups of proteins and the catechol moiety can induce cross-linking of proteins. Altogether, dopamine-derived products have a similar structure and mode of action as MGO and, therefore, have to be taken into consideration as well when discussing the importance of reactive dicarbonyls.
AN INTRIGUING OBSERVATION: ANTI-DIABETIC DRUGS HAVE BENEFICIAL EFFECTS IN PD
Given the commonalities between PD and diabetes, it is tempting to speculate, that anti-diabetic drugs may also prove beneficial in PD. In fact, there are already examples of such drugs.
As mentioned above, diabetes is associated with the accumulation of AGEs. Importantly, reducing glycation is beneficial in models of diabetes. On the other hand, several anti-diabetic drugs act on glycation [84]. Glycation and, therefore, AGE levels, can be modulated in different ways: (i) by reducing the levels of reducing sugars as glucose or dicarbonyls, which consequently reduces AGE formation; (ii) via scavenging of the precursors; (iii) or by enhancing the degradation of dicarbonyls and sugars.
Mitoglitazone (MSDC-016), a drug with anti-diabetic properties that targets the mitochondrial pyruvate carrier, a global controller of cell metabolism, was found to protect against the neurotoxic effects of MPP+ in human cells, and against MPTP in murine, and in nematode models [85]. Remarkably, treatment of mice with mitoglitazone prior to MPTP administration preserved the motor function and reduced dopaminergic cell loss, increasing striatal dopamine levels and reducing neuroinflammation [85]. These findings are consistent with studies in human subjects. In a large retrospective cohort study concluded that diabetic patients treated with the antidiabetic glitazone drugs presented reduced incidence of PD [86]. Moreover, glitazone was also associated with reduced incidence of PD, in comparison with patients treated only with metformin [87]. However, the clinical relevance of these findings is still controversial, as another restrospective cohort study comparing the incidence of PD in diabetes patients found that glitazone was not associated with reduced incidence of PD [88]. Another recent study analysed the effects of glitazone in PD patients and found that it did not modify disease progression [89].
Several other studies suggest that anti-diabetic drugs such as exenatide and metformin are neuroprotective in several models of PD and in patients [90, 91]. Metformin and exenatide both act by lowering the levels of glucose. In addition, metformin can also directly scavenge MGO, giving rise to a non-toxic imidazolinone compound [92].
Importantly, metformin treatment in the MPTP mouse model of parkinsonism improves motor function, protects dopaminergic neurons, and increases levels of the brain derived neurotrophic factor [93–96]. A trial with diabetes patients revealed that Glo1 activity was increased after metformin administration [97].
Telmisartan, an angiotensin II type 1 receptor antagonist used to manage hypertension, was shown to be neuroprotective in MPTP models, reducing degeneration of dopaminergic cells and motor impairment. In addition, it also reduced aSyn accumulation and increased the levels of BDNF [98–100]. Interestingly, telmisartan was also shown to reduce AGE levels in the hippocampus of a rat model of hypertension [98].
Thiamine, also known as vitamin B1, is a cofactor of enzymes involved in glucose metabolism that was suggested to be linked to PD [101, 102]. Intramuscular injection of high-doses of thiamine significantly improved motor function in PD patients [103, 104]. Interestingly, thiamine was shown to reduce AGE levels in rodent models of diabetes [105] and to reduce glucose levels in hyperglycemic patients [106].
Interestingly the MGO-scavengers tenilsetam and aminoguanidine that both alleviate the toxic effects of MGO and were considered as anti-diabetic drugs [107, 108] were shown to reduce aggregation and improve clearance of aSyn in a PD cell model [74]. In addition, the motor performance in a Drosophila PD-model [74].
Collectively, these studies provide evidence that there might be common features between diabetes and PD, and that additional studies are needed [69, 109]. We speculate that some of the variability may come from the fact that the clinical studies have not taken into consideration the different sub-types of PD, and this is something that needs to be accounted for in future trials, to ensure more conclusive results.
CONCLUDING REMARKS
As discussed above, several studies indicate that diabetes patients experience an increased risk to develop PD. However, the underlying molecular mechanisms are still essentially unknown. Interestingly, both diseases are associated with altered sugar metabolism and, therefore, with increased levels of glycation. Glycation is an unavoidable process that causes the formation of AGEs. These are able to induce crosslinks between proteins, which could increase protein aggregation. Since aSyn is a long-lived protein, AGEs might accumulate in aSyn over time, affecting its aggregation and toxicity. Interestingly, AGEs were identified in the periphery of Lewy bodies and aSyn was also found glycated in the brains of PD patients. Glycation was shown to potentiate aSyn associated neurodegeneration in synucleinopathies, suggesting that this could be the underlying molecular mechanism associating diabetes and PD.
Importantly, cells evolved different glycation agents detoxifying mechanisms. Moreover, DJ-1 was suggested to act both as a methylglyoxalase and as a deglycase. This connection further supports the importance of glycation in the etiology of PD.
Given that drugs aiming at decreasing glucose and AGE levels show protective activity in both PD and diabetes, it is tempting to further speculate that glycation may constitute the missing link between PD and diabetes. Formation of AGEs is, for the most part, considered irreversible, which underlines the importance of detecting and treating diabetic conditions at an early stage. Many questions regarding the spontaneous reaction of toxic metabolites with PD-related proteins still pend resolution, but there is enough evidence to spark enthusiasm for future studies.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
Footnotes
ACKNOWLEDGMENTS
HVM is supported by Fundação para a Ciência e Tecnologia (FCT), Portugal (SFRH/BPD/109347/2015; PTDC/NEU-OSD/5644/2014). TFO is supported by the DFG Center for Nanoscale Microscopy of the Brain (CNMPB) and by an EU Joint Programme – Neurodegenerative Disease Research (JPND) project (aSynProtec). The project is supported through the following funding organisations under the aegis of JPND – ![]() (BMBF).
(BMBF).