
Abstract
α-synuclein is a small protein abundantly expressed in the brain and mainly located in synaptic terminals. The conversion of α-synuclein into oligomers and fibrils is the hallmark of a range of neurodegenerative disorders including Parkinson’s disease and dementia with Lewy bodies. α-synuclein is disordered in solution but can adopt an α-helical conformation upon binding to lipid membranes. This lipid-protein interaction plays an important role in its proposed biological function, i.e., synaptic plasticity, but can also entail the aggregation of the protein. Both the chemical properties of the lipids and the lipid-to-protein-ratio have been reported to modulate the aggregation propensity of α-synuclein. In this review, the influence of changes in the nature and levels of lipids on the aggregation propensity of α-synuclein in vivo and in vitro will be discussed within a common general framework. In particular, while biophysical measurements and kinetic analyses of the time courses of α-synuclein aggregation in the presence of different types of lipid vesicles allow a mechanistic dissection of the influence of the lipids on α-synuclein aggregation, biological studies of cellular and animal models of Parkinson’s disease allow the determination of changes in lipid levels and properties associated with the disease.
Keywords
α-synuclein is a small intrinsically disordered protein mainly found in pre-synaptic terminals [1] and whose aggregation has been associated with a range of neurodegenerative disorders, such as Parkinson’s disease (PD) and dementia with Lewy bodies (DLB) [2]. In particular, α-synuclein was found to be the main constituent of Lewy bodies which are composed of proteinaceous aggregates found in the brains of patients with PD [3]. Although the exact function of α-synuclein has not yet been defined, the protein is proposed to interact with biological membranes and membrane proteins and to play a role in synaptic plasticity and neurotransmitter release [4]. In particular, α-synuclein was found to promote the assembly of the SNARE complex in vivo and in vitro via the formation of a multimer at the surface of synaptic vesicles [5]. These vesicles are small organelles, with a diameter of ~40 nm, and are composed of proteins and lipids at a protein to phospholipid mass ratio of 2 [6]. The main constituting lipids of synaptic vesicles include cholesterol, phospholipids (phosphatidylcholine (32%), phosphatidylserine (12%), phosphatidylethanolamine (54%), phosphatidylinositol (2%)) and sphingomyelin [6]. The lipid composition of biological membranes varies from one organelle to another [7] and the constituting lipid molecules mainly vary in the nature of their hydrophobic hydrocarbon chain(s) (e.g.number, length and degree of unsaturation) and their polar head (e.g., charged, zwitterionic, glycosylated) [8] (see representative structures in Fig. 1).
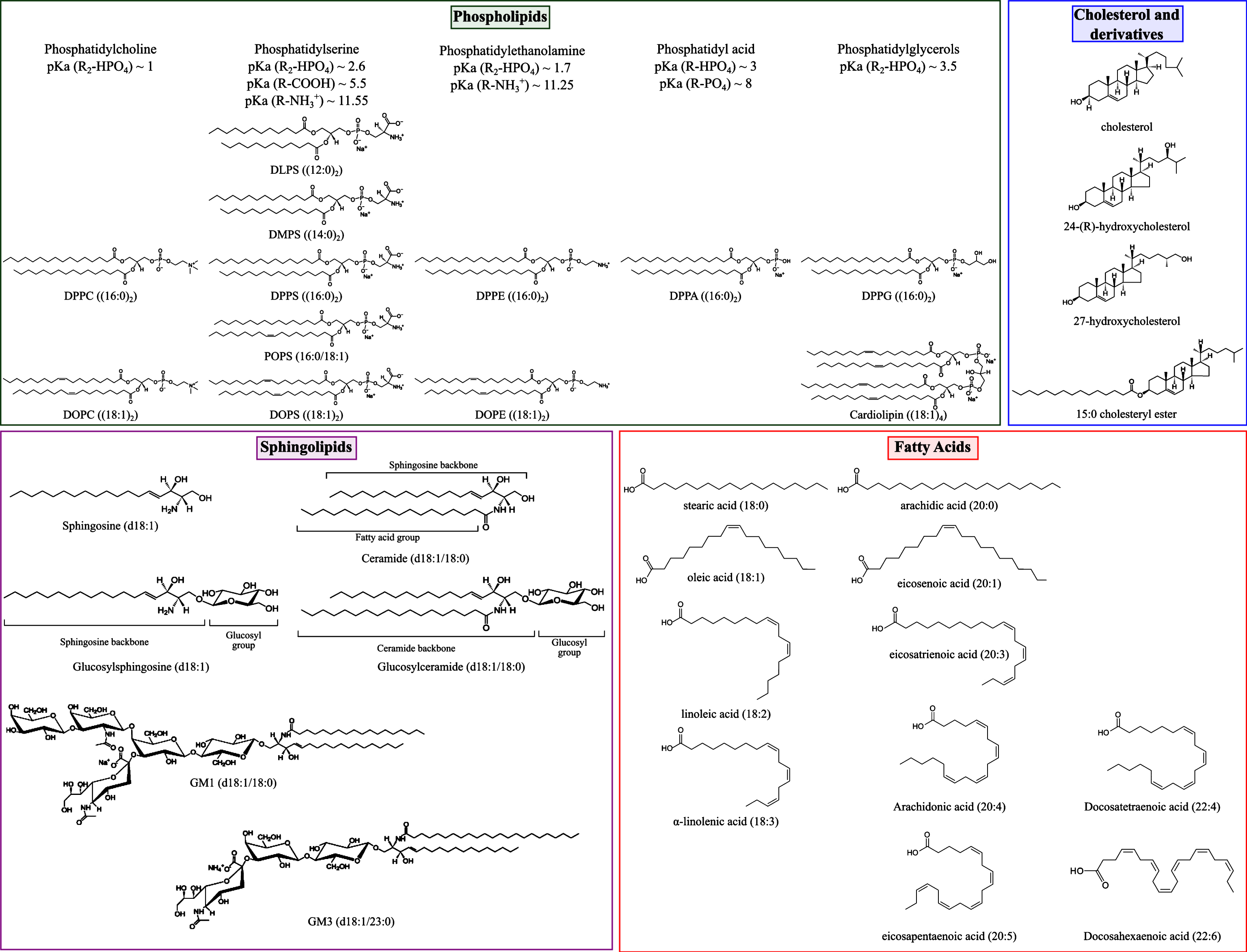
Representative structures of lipid molecules found to influence the aggregation of α-synuclein in vitro and in vivo (see main text). DLPS: 1,2-dilauroyl-sn-glycero-3-phospho-L-serine; DMPS: 1,2-dimyristoyl-sn-glycero-3-phospho-L-serine; DPPC: 1,2-dipalmitoyl-sn-glycero-3-phosphocholine; DPPA: 1,2-dipalmitoyl-sn-glycero-3-phosphate; DPPG: 1,2-dipalmitoyl-sn-glycero-3-phospho-(1’-rac-glycerol); DPPE: 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine; DPPS: 1,2-dipalmitoyl-sn-glycero-3-phospho-L-serine; POPS: 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine; DOPS: 1,2-dioleoyl-sn-glycero-3-phospho-L-serine; DOPE: 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine; DOPC: 1,2-dioleoyl-sn-glycero-3-phosphocholine; cardiolipin: 1,3-bis[1,2-dioleoyl-sn-glycero-3-phospho]-sn-glycerol. pKa values for phospholipids are indicated for an ionic strength of 0.1M NaCl [56].
Upon binding to membranes, a fraction of the first 95 residues of α-synuclein undergoes a structural transition from random coil to α-helix and the proportion of the sequence involved in this transition strongly depends on the overall lipid-to-protein-ratio [9, 10] and on the nature of the lipids constituting the membrane [11]. Molecular dynamics (MD) simulation, nuclear magnetic resonance (NMR) and neutron reflectometry studies have shown that the amphipathic helix formed by α-synuclein sits between the polar groups of adjacent lipids at a depth corresponding to that of the glycerol group [12–14]. In agreement with this model, α-synuclein binds less favourably to multi lamellar vesicles (MLV) than to small unilamellar vesicles (SUV) for the same lipid concentration because fewer lipid molecules are exposed at the surface of MLV compared to that of SUV [15, 16]. The interaction between α-synuclein and membranes was found to affect not only the properties of the protein but also those of the membrane. Indeed, the binding of α-synuclein to membranes can induce a change in their melting temperature [17–19], membrane remodelling [20–25], membrane thinning and membrane expansion [24, 26].
It is now well established that the physical, chemical and thermotropic properties of the membrane affect the strength of the binding of α-synuclein and that the protein binds preferentially to membranes composed of negatively charged lipids and/or containing lipid packing defects [26, 27] and/or in the fluid phase [18, 19]. In addition, it has been shown that a change in the ionic strength [15, 28–31] or a decrease of the ionic density at the surface of the membrane [19, 29] led to a decrease of the strength of α-synuclein lipid binding, emphasising the importance of electrostatics in the α-synuclein-lipid interaction. Moreover, the introduction of cationic molecules [32] or other lipid binding proteins [33] can interfere with the binding of α-synuclein to membranes via competition for binding sites at the surface of the vesicles.
The equilibrium between the unfolded and α-helical membrane bound states of α-synuclein has also been found to modulate its propensity to aggregate both in vivo and in vitro [34]. Although the structural details of the binding between α-synuclein and lipid membranes have been well characterised [9, 11], the mechanisms by which lipid vesicles influence the aggregation of α-synuclein both in vivo and in vitro are not yet well understood. Over the last two decades, a large range of biological and biophysical studies have been performed, aimed at elucidating the relationship between the nature of the interaction of α-synuclein with lipids and the propensity of the protein to aggregate [19, 34–42]. Indeed, changes in the levels of specific lipids, including fatty acids, sphingolipids, cholesterol derivatives (seerepresentative structures in Fig. 1), have been associated with increased levels of soluble and aggregated α-synuclein in different cellular and animal models of PD and associated disorders [41, 43–48]. In parallel to these biological studies, the influence of a large number of lipids on the kinetics of amyloid formation by α-synuclein was investigated in vitro using recombinant protein and synthetic lipids [19, 49] (Fig. 1). The magnitude by which lipids affect the aggregation of α-synuclein was found to be related to the strength of the binding of α-synuclein to lipids and mainly two parameters: the lipid-to-protein-ratio (L:P ratio) and the chemical properties of the lipids [19, 38]. In addition, the exposure of α-synuclein to certain lipids was found both in cell cultures and in vitro to lead to the formation of a variety of mixed protein-lipid structures, in the form of either lipoprotein particles [50, 51], oligomers [38, 52–54], amyloid fibrils [55] and clusters at the surface of membranes [56–58] (Fig. 2). Moreover, the nature of the structures formed by α-synuclein highly depends on the L:P ratio(Fig. 2).
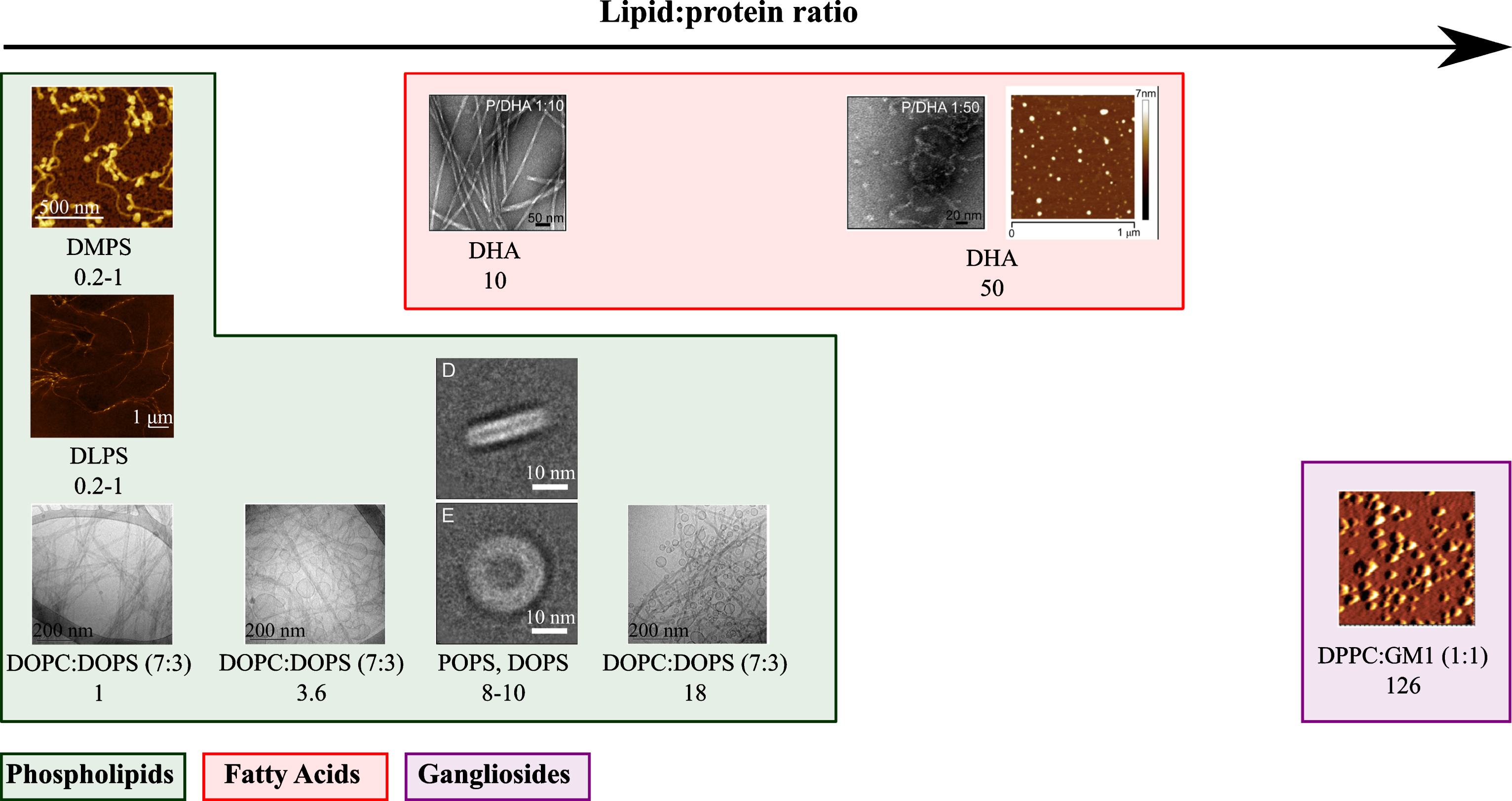
Representative structures that α-synuclein can form when the recombinant protein was incubated in the presence of lipids at different L:P ratios. For each protein-lipid system, the L:P (M:M) ratio and the lipid composition are indicated. The atomic force microscopy and electron microscopy images were adapted from [19, 55].
In this review, the role of lipids on the kinetics of amyloid formation by α-synuclein will be reviewed in the context of the pathogenesis of PD. First, the effect of lipids on the mechanism and kinetics of α-synuclein aggregation in vitro will be discussed for two sets of conditions: aggregating and non-aggregating conditions. Then, the influence of changes in the levels of specific lipids on those of aggregated α-synuclein in cellular and animal models of PD will be discussed. The effect of lipids on the aggregation of α-synuclein in cell cultures and in vivo will be discussed in relation to the findings obtained from in vitro experiments.
ROLES OF LIPIDS ON THE AGGREGATION OF α-SYNUCLEIN IN VITRO
In this section, the influence of lipid vesicles on the different steps of α-synuclein aggregation will be discussed for different sets of conditions. In most cases, the aggregation of α-synuclein is followed through increases in Thioflavin-T (ThT) fluorescence. ThT binds only to β-sheet-rich fibrillar protein, and not to monomers, and its enhanced fluorescence upon binding to amyloid fibrils can be used to detect and quantify the amount of amyloid fibrils present. An increasing body of experimental evidence shows that soluble, monomeric α-synuclein is kinetically stable (’metastable’) at physiological concentration(∼ 50μM [59]) and neutral pH for several weeks at room temperature in the absence of catalytic surfaces. Aggregation experiments with α-synuclein are therefore often performed under conditions of mechanical agitation (’shaking’). Under such conditions, the spontaneous aggregation of α-synuclein is dramatically accelerated, most likely through a combination of enhanced turnover of aggregate species that form at the air-water interface [60], as well as enhanced fragmentation of fibrils, which leads to an exponential increase in aggregation rate [61]. Other surfaces, such as uncoated polystyrene surfaces of multiwell plates [62, 63] or polymeric nanoparticles [64] also are able to induce the primary nucleation of α-synuclein amyloid fibrils. If, furthermore, the solution pH is mildly acidic (pH < 6), autocatalytic secondary nucleation is able to exponentially amplify small numbers of aggregates [63, 65]. We will from now on refer to the set of conditions under which α-synuclein shows spontaneous aggregation within the typical time scale of an in vitro aggregation experiment (1 week) as ‘aggregating conditions’, whereas we refer to conditions under which no aggregation can be detected by means of the ThT assay within such a time scale as ‘non-aggregating conditions’ (Fig. 3). Both of these types of conditions have been used in the context of α-synuclein lipid studies. While aggregating conditions are particularly useful to explore the ways in which lipids can modulate the overall aggregation of α-synuclein, non-aggregating conditions can be used to determine whether or not a given type of lipid can induce the de novo aggregation of α-synuclein[19, 35].
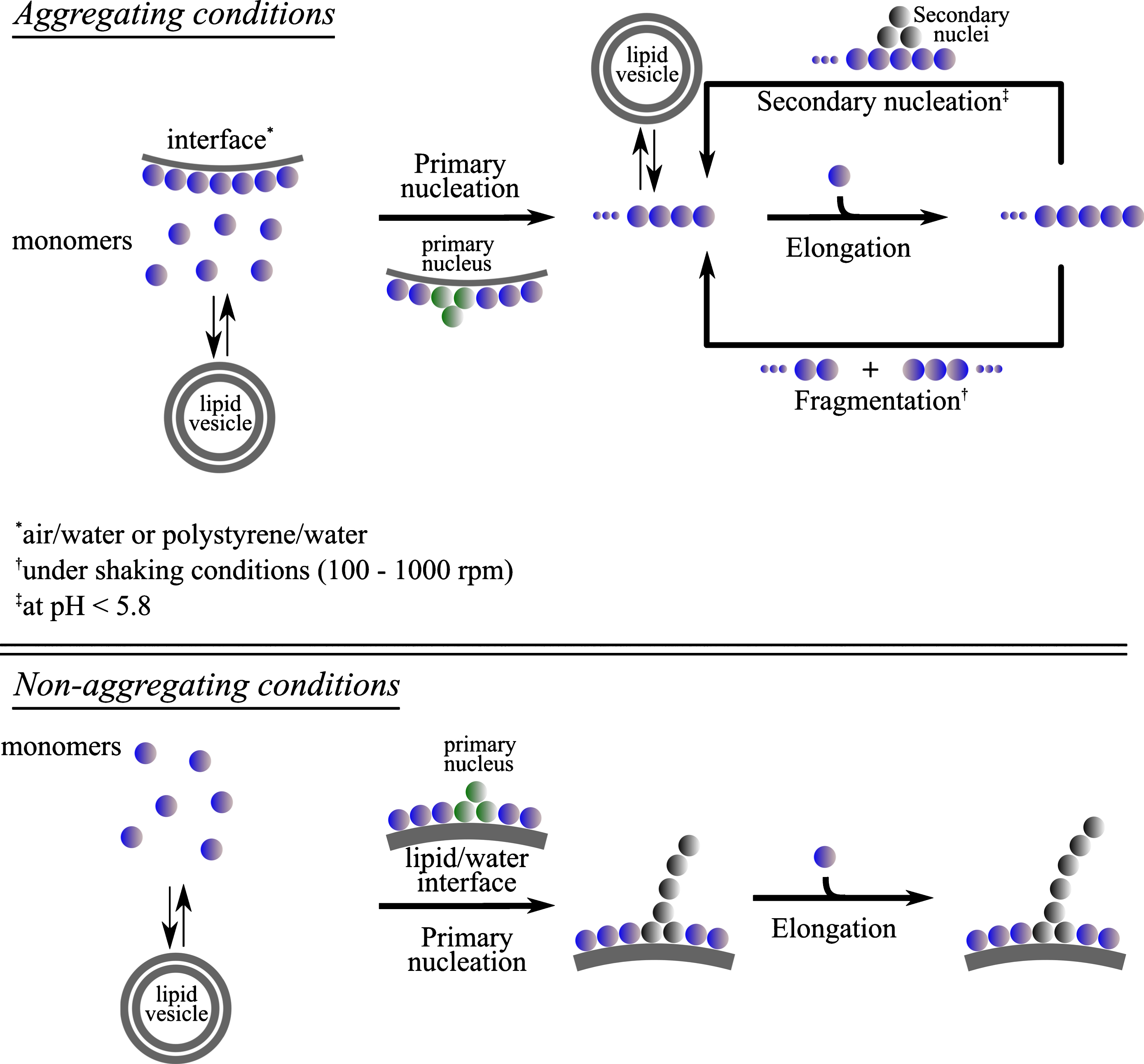
Proposed networks for the aggregation reaction of α-synuclein in the presence of lipid vesicles under quiescent and shaking conditions. The presence of lipid vesicles was found to influence different steps of α-synuclein aggregation, primary and secondary nucleation, as a result of interactions between the vesicles and the monomeric [28, 37] or fibrillar [35, 55] state of the protein, respectively.
Under aggregating conditions, the observed effect of lipids on the kinetics of α-synuclein aggregation can therefore be due to the perturbation of one or several of the individual steps via interactions of lipids with one or more of the species along the reaction network, e.g. monomers, primary and/or secondary nuclei, fibrils (Fig. 3), as it has been postulated for the effect of chaperones on the kinetics of protein aggregation [66].
The magnitude by which lipid systems either accelerate or inhibit the reaction of amyloid fibril formation was found to correlate with the affinity of monomeric α-synuclein for these lipids [19, 37]. Indeed, lipid systems, with which α-synuclein was found not to favourably interact, did not influence the rate of its aggregation (ratio thalf ∼ 1, whereby thalf is the time it takes for 50% of the available protein monomer to be converted into amyloid fibrils) (see Table 1) [28, 37]. However, lipid systems containing negatively charged phospholipids, including phosphatidylethanolamine (PE), phosphatydic acid (PA), phospho-glycerol (PG), phosphatidylserine (PS), sphingolipids or fatty acids, were found to initiate, accelerate or inhibit α-synuclein aggregation [19, 49].
Kinetics of aggregation and lipid binding properties of α-synuclein in the presence of model membranes in vitro
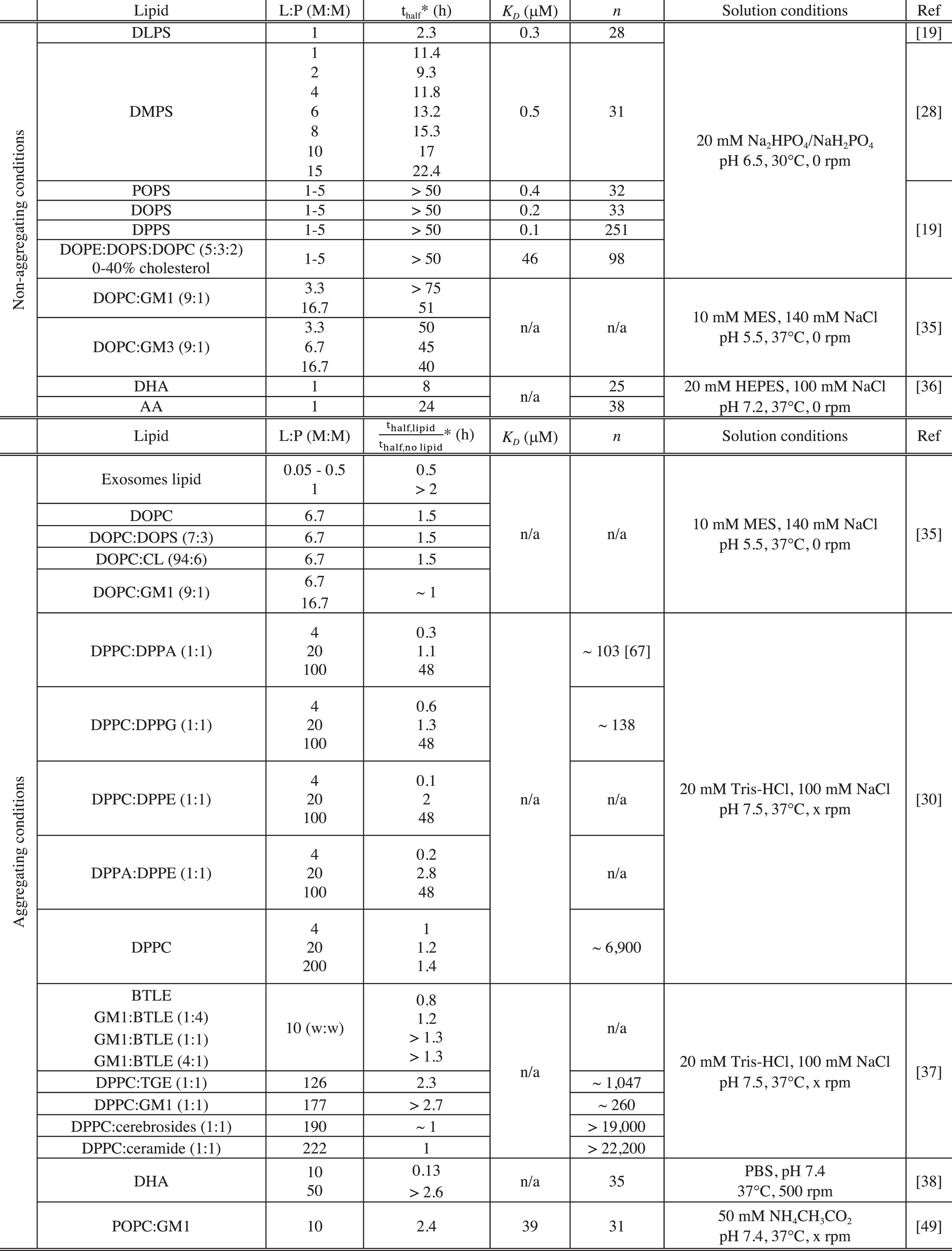
PBS: Phosphate-buffered saline, pH 7.4; GM1: GM1 Ganglioside (Brain, Ovine); GM3: GM3 Ganglioside (Milk, Bovine); CL: cardiolipin; BTLE: Brain Total Lipid Extract (Porcine); TGE: Total Ganglioside Extract (Brain, Porcine); Cerebrosides: Total Cerebrosides (Brain, Porcine); Ceramide: Ceramide (Brain, Porcine); DHA: docosahexaenoic acid (22:6(ω-3)); AA: arachidonic acid (20:4(ω - 6)); n/a: not available. *For the data acquired under non-aggregating conditions, we reported the value of the observed half-time (thalf) of the reaction of amyloid formation by α-synuclein in the presence of a given lipid system. For the data acquired under aggregating conditions, we indicated the value of the ratio
Negatively charged phospholipids
The magnitude by which lipid vesicles influence the kinetics of α-synuclein aggregation was found to be strongly dependent on the L:P ratio and this observation can be rationalised by considering the availability of soluble monomeric α-synuclein and of α-synuclein bound to lipid vesicles. Indeed, it is possible to evaluate the concentration of protein free in solution and bound to the vesicles for any given L:P ratio, if the binding constant, K D and stoichiometry, n (number of lipid molecules bound to one molecule of α-synuclein), have been characterised for a given protein-lipid system (see Table 1). This rationalisation of the effects of lipids will first be illustrated using kinetic measurements performed under non-aggregating conditions (Fig. 4) and then be used to describe and explain the kinetic data measured under aggregating conditions.
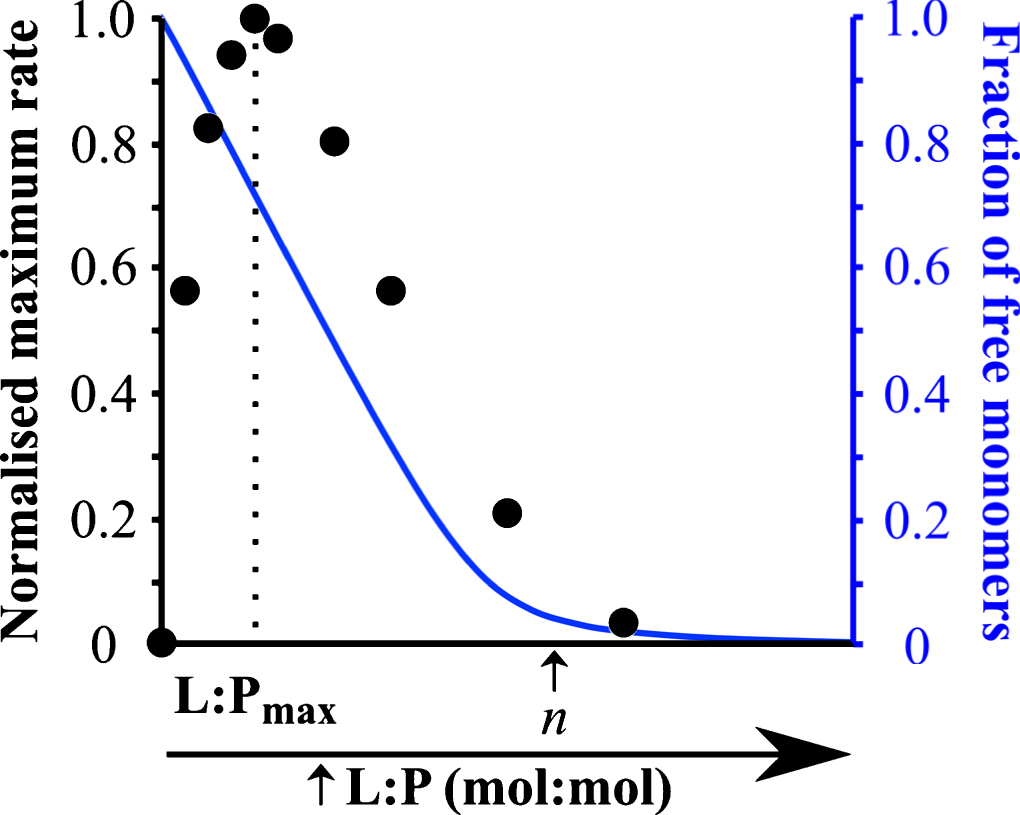
Influence of the L:P ratio on the kinetics of α-synuclein aggregation. This plot shows (i) the rate of α-synuclein aggregation (black) and (ii) the fraction of soluble protein (blue) in the presence of different DMPS: α-synuclein ratios. Figure adapted from [28].
Under quiescent conditions and at neutral pH, α-synuclein does not form detectable amounts (by ThT fluorescence) of amyloid fibrils, because the rate of homogeneous primary nucleation is very slow. Using such conditions, it is possible to quantitatively determine the role of lipid vesicles on α-synuclein aggregation. At L:P ratios < n, the presence of certain negatively charged lipid vesicles (made from DLPS and DMPS) induces detectable aggregation under otherwise non-aggregating conditions by enhancing the rate of its primary nucleation by several orders of magnitude [28] (Fig. 4). At these L:P ratios, both protein free in solution and protein bound to the membrane are present and can contribute to the reaction of amyloid formation [19, 28] (Fig. 4). In particular, the ThT assay requires significant amounts ofamyloid fibrils to be formed (of the order of 1 μM [28]), which in turn requires a sufficiently high concentration of free monomer that enables the nuclei, that are formed at the lipid surface, to grow. At L:P > n, however, α-synuclein does not form amyloid fibrils because all the soluble monomers have been depleted from the solution due to their adsorption at the surface of the vesicles [28] (Fig. 4). For intermediate L:P ratios (0 < L:P < n), the rate of α-synuclein aggregation first increased with increasing L:P ratios to reach a maximum value for a L:P ratio, L:Pmax (Fig. 4). Under these conditions, a large excess of free monomers of α-synuclein are present in solution and increasing the number of lipid vesicles results in an increase in the number of nucleation sites and thus faster kinetics of aggregation (Fig. 4). However, for L:P ratios > L:Pmax, the rate of aggregation decreased and eventually reached undetectable levels, because the concentration of soluble monomeric α-synuclein, that is available for the growth of the nuclei, decreases with increasing L:P ratios due to the adsorption of protein molecules at the surface of the vesicles [28] (Fig. 4).
The same rationale can be applied to explain the modulation, by lipids, of the kinetics of amyloid formation of α-synuclein under aggregating conditions (shaking and neutral pH and/or untreated polystyrene plates). In particular, below a L:P ratio of 4, the presence of DPPC:DPPA (1:1) and DPPC:DPPG (1:1) vesicles was found to accelerate the reaction of amyloid formation [30].
Under these conditions, both protein free in solution and bound to the vesicles are present and the lipid vesicles are likely to influence the aggregation process by providing an additional pathway of heterogeneous primary nucleation, as observed for DMPS and DLPS vesicles [19, 28]. However, for higher L:P ratios, these vesicles were found to slow down the reaction of amyloid formation until complete inhibition was observed at L:P > n. Furthermore, α-synuclein was found to form lipoprotein particles (∼25 nm diameter) when incubated with DOPS and POPS under aggregating conditions at L:P ratio > n. The mixed DOPS-α-synuclein particles were composed of 8 molecules of α-synuclein and 1,070 molecules of DOPS and the protein mainly adopts an α-helical conformation within these structures [50].
Altogether, these results suggest that the presence of negatively charged vesicles at L:P ratio < n and under neutral conditions can trigger or accelerate α-synuclein aggregation most likely by providing an alternative route for primary nucleation. However, at L:P ratios > n, α-synuclein does not form significant amounts of amyloid fibrils but is mainly associated with lipids in a predominantly α-helical, thermodynamically stable conformation.
Under acidic pH conditions, the presence of vesicles made from DOPC, DOPC:DOPS (70:30) and DOPC:CL (95:5) were found to decrease the rate of α-synuclein aggregation at a L:P ratio of 6.7 [35]. Under these conditions, both DOPC and DOPC:DOPS (70:30) vesicles were found (from analysis of cryo-electron microscopy) to bind to the sides of the fibrils [55]. It is important to note that, at this slightly acidic pH, secondary nucleation, i.e. the autocatalytic formation of new aggregates on the surface of existing fibrils, also contributes to the overall reaction of amyloid formation [63, 65] (Fig. 3).
The observed decrease in the rate of aggregation could therefore result from the binding of lipid vesicles to the surface of the fibrils, thus preventing the access of monomeric α-synuclein to the catalytic secondary nucleation sites. In addition to the L:P ratio, the chemical properties of lipids were also found to influence the magnitude by which they can initiate the aggregation of α-synuclein [19]. Indeed, although α-synuclein binds with similar affinity to different negatively charged lipid vesicles (DOPS, POPS, DOPE:DOPS:DOPC (50:30:20, w:w:w), DMPS and DLPS), the protein was found to aggregate only in the presence of those vesicles made from lipidscharacterised by relatively high solubility in water (DLPS, DMPS) [19].
This suggests that the chemical environment of at least some of the lipid molecules is changed during the aggregation process, a hypothesis in agreement with the reported formation of mixed protein lipid structures [55].
All together, these results suggest that the L:P ratio influences both the kinetics of amyloid formation by α-synuclein and the nature of the aggregates formed in the presence of vesicles made from negatively charged phospholipids (see Fig. 2). In addition, both monomeric and fibrillar species of α-synuclein can bind to lipid vesicles and the latter can thus influence different steps along the aggregation pathway (i.e., primary and secondary nucleation).
Sphingolipids
Vesicles containing sphingolipids (Fig. 1) were also found to affect the kinetics of α-synuclein amyloid formation under aggregating conditions at neutral pH [37]. The chemical properties of the sphingolipids were found to influence the efficiency of the binding of monomeric α-synuclein and as a consequence the magnitude by which they can affect the rate of α-synuclein amyloid formation [37] (see Table 1). Indeed, α-synuclein was found not to interact with vesicles made from DPPC:cerebrosides (1:1), DPPC:ceramides (1:1) and their presence did not affect the kinetics of amyloid formation by α-synuclein under aggregating conditions (with mechanical agitation) [37] (Table 1). However, the protein was found to bind to DPPC:total gangliosides (1:1) (n ∼ 1,000) and to a greater extent to DPPC:GM1 (1:1) (n ∼ 200) and the presence of these vesicles at a L:P ratio of 130 and 180, respectively, were found to either slow the reaction down or to completely inhibit it, respectively [37]. At these L:P ratios, a fraction of α-synuclein molecules is bound to the DPPC:total gangliosides (1:1) vesicles leading to a decrease of the overall rate of aggregation whereas most of α-synuclein molecules are bound to the DPPC:GM1 (1:1) vesicles leading to the absence of detectable amyloid formation, under these conditions. It is interesting to note that the presence of DPPC:total gangliosides (1:1) mainly affects the lag phase of the aggregation time course, suggesting that these vesicles are likely to influence the primary nucleation step, a conclusion that can only be drawn in the absence of secondaryprocesses [66].
As observed for DLPS and DMPS (see section ‘Negatively charged phospholipids’), vesicles made from lipids characterised by relatively high solubility in water (GM1 and GM3) were found to be able to initiate the reaction of amyloid formation under acidic pH and quiescent conditions [35]. It is difficult, however, to compare the magnitude by which the gangliosides containing vesicles and the DLPS and DMPS vesicles can initiate the aggregation of α-synuclein as the kinetics of amyloid formation were reported under two different sets of solution conditions (pH 5.5 for the gangliosides vs. pH 6.5 for the PS lipids). As mentioned above, under mildly acidic conditions (pH < 6), an additional reaction step, the secondary nucleation which induces the exponential multiplication of fibrils, also contributes to the overall rate of amyloid formation [65, 63]. In the presence of this amplification pathway, even a very low rate of primary nucleation (i.e. de novo formation of amyloid fibrils) can lead to rapid aggregation, rendering accurate determination of the primary nucleation rate difficult. Taken together, these results suggest that the solubility of a given lipid is likely to be one of the key properties contributing to its ability to initiate α-synuclein aggregation.
Although the presence of vesicles made from DOPC:GM1 or DOPC:GM3 were able to initiate α-synuclein aggregation under quiescent conditions, these vesicles were found not to affect the kinetics of α-synuclein aggregation at acidic pH under conditions of mechanical agitation at the same L:P ratios [35]. This observation can be explained by considering the different steps involved in the reaction of amyloid formation under these two solution conditions. Indeed, the experimental details of the study under quiescent conditions suggest that the first step of the aggregation in passivated (’non binding’) multi-well plates is the primary nucleation at the surface of the vesicles. However, in the case of the study under acidic pH and shaking conditions in non-passivated multi-well plates, the primary nucleation is likely to occur at the polystyrene surface of the plates [63]. These results suggest that the presence of vesicles does not affect the overall rate of α-synuclein aggregation in polystyrene plates and thus that the rate of primary nucleation at the polystyrene/water interface is likely to be faster than that of the primary nucleation at the vesicle/water interface. Therefore, the solution conditions used in studies of the role of lipid vesicles in the initiation of the aggregation of α-synuclein need to be carefully controlled in order to minimise other contributions that can trigger the aggregation of the protein and that can mask the contribution of lipid vesicles.
Fatty acids
Unlike lipid molecules mentioned in the previous two paragraphs, fatty acids (FA (C:D)) are carboxylic acids characterised by only one hydrocarbon chain of a given length, C (number of carbons) and either saturated (no double bonds/unsaturations, D = 0) (SFA), monounsaturated (one unsaturation, D = 1) (MUFA) or polyunsaturated (more than one unsaturation, D > 1) (PUFA) (see representative structures in Fig. 1). PUFA are particularly susceptible to oxidative stress as their double bonds are particularly reactive towards oxidative radicals [68].
Under quiescent conditions, the presence of certain FA such as arachidonic acid (AA) (20:4) and docosahexaenoic acid (DHA) (22:6) were found to initiate the aggregation of α-synuclein when they were incubated at a L:P ratio of 1 [36]. In addition, DHA was found to accelerate the reaction of amyloid formation by α-synuclein under conditions of mechanical agitation at a L:P ratio of 10 [38].
Given that the number of lipid molecules bound to a molecule of α-synuclein is 25 and 38 for DHA and AA, respectively [36] (Table 1), both the protein free in solution and bound to the FA are populated and can contribute to the reaction of amyloid fibril formation, at these L:P ratios. Moreover, the presence of DHA is likely to provide an alternative route for primary nucleation as it was found to be able to initiate the aggregation of α-synuclein under quiescent conditions.
At higher L:P ratios (L:P > n), the incubation of α-synuclein with FA led to the formation of multimers under both quiescent and shaking conditions [38, 69]. In particular, α-synuclein was found to form dimers and trimers when incubated with AA (L:P = 1000) under quiescent conditions [54]. In addition, the presence of AA (L:P ~30), DHA (L:P > 50) or α-linolenic acid (ALA) (18:3) (L:P = 1000) was found to induce the formation of oligomers by α-synuclein under shaking conditions [38, 69]. Moreover, the AA- and DHA-α-synuclein oligomers were not able to convert into fibrils [38, 69] and the presence of DHA at this L:P ratio led to the inhibition of the reaction of amyloid formation by α-synuclein [38]. Furthermore, AA and DHA were found to form covalent adducts with α-synuclein, the formation of which was found to protect AA and DHA from oxidative damage [70]. At these L:P ratios, all the molecules of α-synuclein are bound to the FA, which is likely to favour the formation of mixed protein-lipid structures as observed upon incubation of α-synuclein and DOPS or POPS under shaking conditions and at high L:P ratio [50] (see section ‘Negatively charged phospholipids’). These observations suggest that the observed inhibition of α-synuclein amyloid formation by FA at high L:P ratios may be rationalised by the formation of off-pathway mixed FA-protein oligomers, that are thermodynamically more stable than amyloid fibrils under these conditions.
Finally, the toxicity of FA induced oligomers to neuronal cells was found to differ: the AA-induced α-synuclein oligomers were found not to cause ROS production in primary neuronal cultures [69] whereas the DHA-induced α-synuclein oligomers are toxic to SH-SY5Y cells and permeabilise PG membranes [52]. These results suggest that the chemical properties of the FA are likely to influence the intrinsic characteristics of the mixed FA-α-synuclein oligomers, e.g. hydrophobicity, and, as a result, their toxicity to cells.
INTERPLAY BETWEEN THE LEVELS OF LIPIDS AND THOSE OF α-SYNUCLEIN IN CELLULAR AND ANIMAL MODELS OF PARKINSON’S DISEASE
Changes in the levels of both specific lipids and α-synuclein have been associated with PD [41, 43–46]. In particular, these changes have been identified in different cellular and animal models of the disease, including human neuroblastoma cell lines, human iPSC-derived dopaminergic neurons and post-mortem human and rodent brains [41, 43–46] (Table 2). In these models, the processes associated with PD were reproduced mainly via exposure to toxins [41, 72] or modulation (e.g., mutations, knock-down or knock out) of genes encoding either α-synuclein [46] or proteins associated with the disease [41, 45], leading to a higher propensity of α-synuclein to oligomerize/aggregate or the inhibition/enhancement of the activity of the corresponding protein, respectively. Both cellular and animal models of PD were used to recapitulate the pathology associated with the disease and changes in the levels of mainly three classes of lipids were reported: fatty acids, sterols and sphingolipids.
Interplay between the levels of lipids and those of α-synuclein in cellular and animal models of Parkinson’s disease
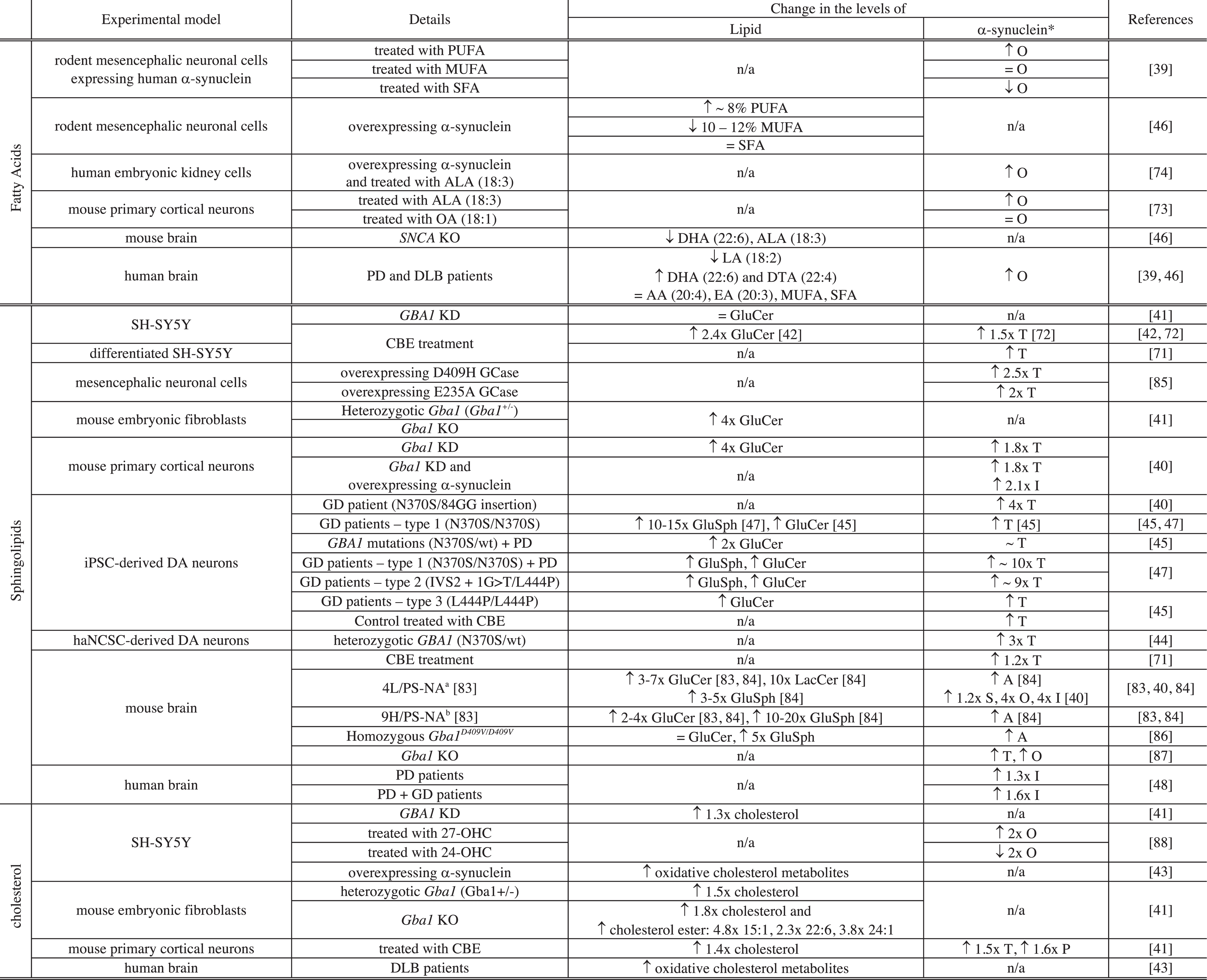
Notes related to Table 2: *level of protein expressed as T: total level of α-synuclein, S: soluble, O: oligomers, I: insoluble, P: pathologic (phosphorylated α-synuclein (S129)); PUFA: polyunsaturated FA; MUFA: monounsaturated FA; SFA: saturated FA; SNCA: gene encoding α-synuclein; KO: knock-out; KD: knock-down; ALA: α-linolenic acid; LA: linoleic acid; DTA: docosatetraenoic acid; EA: eicosatrienoic acid; OA: oleic acid; GluSph: Glucosylsphingosine; iPSC-derived DA neurons: dopaminergic (DA) neurons derived from induced pluripotent cells derived from skin fibroblasts of patients with Gaucher’s (GD) and/or Parkinson’s disease (PD) or healthy controls; haNCSC-derived DA neurons: DA neurons derived from human adipose Neural Crest Stem Cells; a4L/PS-NA = V394L/V394L + PS-/-+low-level mouse prosaposin transgene expression. b9H/PS-NA = D409H/D409H + PS-/- +low-level mouse prosaposin transgene expression.
In this section, changes in the levels of lipids, which were found to be associated with either a gain of toxicity or a loss of function of α-synuclein, will be discussed. The aggregation propensity of α-synuclein in cellular and animal models of PD and in the brain of PD patients associated with a change in the level of a given lipid will be related to that observed in the presence of synthetic vesicles made from this lipidin vitro, when possible.
Fatty acids
Changes in the levels of FA have been associated with changes in the level of α-synuclein in both cellular and animal models of PD and in patient tissues (Table 2) [39, 74]. Polyunsaturated FA (PUFA) (number of unsaturation > 1) are important for neuronal membranes as they can affect their fluidity and permeability and they are proposed to serve as energy reservoir and to take part in intra- and extracellular signalling as second messengers [68]. PUFA are also involved in generation of apoptotic signals, cellular proliferation and translocation of lipid-modifying enzymes to cellular membranes [68].
Treatment of rodent mesencephalic neuronal (MES) cells or primary cortical neurons with PUFA was found to increase the level of oligomers of α-synuclein in the cytosol and the extent by which the level of oligomers was increased correlates with the degree of unsaturation of the FA (α-linolenic acid (18:3) > oleic acid (18:1) > stearic acid (18:0); eicosapentaenoic acid (20:5) > eicosenoic acid (20:1) > arachidic acid (20:0)) [39, 73]. On the contrary, the treatment of these cells with SFA (e.g. stearic acid (18:0) and arachidic acid (20:0)) or MUFA (e.g., oleic acid (18:1) and eicosenoic acid (20:1)), led to either a decrease or no change, respectively, of the level of α-synuclein oligomers in the cytosol [39, 73]. Conversely, when MES cells were transfected with α-synuclein, the total cytosolic content of PUFA was found to increase by 8% whereas those of MUFA and SFA were found either to decrease by 10–12% or not to be affected, respectively [46]. In addition, the treatment of Human Embryonic Kidney cells (HEK293) expressing α-synuclein with α-linolenic acid (18:3) induced an increase in the level of oligomerisation of the protein compared to untreated cells [74]. The interplay between the nature of accumulated FA and the level of α-synuclein oligomers was also observed in mouse and human brains [46]. Indeed, the deletion of the α-synuclein gene in mice led to a decrease in the level of DHA (22:6) and ALA (18:3) [46]. In addition, the level of α-synuclein oligomers was found to increase in the brain of PD and DLB patients and this increase has been associated with i) a decrease in the level of linoleic acid (18:2), ii) an increase in the level of DHA (22:6) and docosatetraenoic acid (22:4) and iii) no change in the level of AA (20:4), eicosatrienoic acid (20:3), MUFA and SFA [39, 46]. Although changes in the level of AA have not been associated with a gain of toxicity of α-synuclein, the interaction betweenα-synuclein and AA can lead to a loss of function of the SNARE complex [75]. Indeed, even though α-synuclein was not found to directly interact with the SNARE complex, the protein was reported to affect the formation of the complex both in vitro and in vivo by sequestering AA, which is then no longer available for the assembly of the complex and its exocytosis [75]. This observation suggests that an increase in the level of α-synuclein, e.g., via SNCA gene duplication and triplication [76, 77], that has been associated with early onset of PD, can also lead to a loss of function of protein partners [78].
Taken together, these results suggest that the nature of FA influences the propensity of α-synuclein to oligomerize in neuronal cell cultures and in vivo. In particular, the levels of most PUFA were found to be directly correlated to those of α-synuclein and the magnitude by which increased levels of PUFA led to increased levels of oligomeric α-synuclein was proportional to the degree of unsaturation and length of their hydrocarbon chain. These results are in agreement with those obtained in vitro using recombinant α-synuclein incubated in the presence of synthetic FA (see section ‘
Sphingolipids
The occurrence of mutations in the gene of the lysosomal enzyme glucocerebrosidase, GCase (GBA1), is the most important risk factor associated with PD [79]. GCase is responsible for the conversion of glucosylceramide (GluCer) to glucose and ceramide in the lysosome and a decrease in its activity was found to lead to the accumulation of its substrates, e.g. GluCer and glucosylsphingosine (GluSph), and compromise lysosomal activity[40, 81]. The deficiency in GCase activity was also found to impair lysosomal recycling leading to accumulation of defective lysosomes unable to proceed to autophagic clearance of α-synuclein leading to an increase in the levels of the protein [41]. Indeed, accumulation of GluCer and GluSph and increased levels of α-synuclein were found to be associated with GCase deficiency both in mammalian cells and in vivo (mouse and human) [40, 82–84] (see Table 2). In both cellular and animal experimental models, GCase deficiency was due to either chemical treatment with an inhibitor of GCase (Conduritol-B-epoxide (CBE)) [41, 72], GBA1 mutations (E235A, N370S, V394L, D409H or D409V) [40, 85], GBA1 knockdown (KD) [40, 41] or GBA1 knock-out (KO) [41] (Table 2).
Although, deficiency of GCase has been associated with increasing levels of GluCer, GluSph and total soluble α-synuclein, it was not always associated with the presence of detectable insoluble or pathological α-synuclein (Table 2). Indeed, the presence of these species was mainly observed in the brain of GD mouse models (4L/PS-NA, 9H/PS-NA and homozygous Gba1 D409V/D409V), in Gba1 KD mouse primary cortical neurons overexpressing α-synuclein and mouse primary cortical neurons treated with CBE [40, 86].
Interestingly, the activity of GCase could be restored by treating (i) iPSC-derived macrophages and dopaminergic neurons from patients with Gaucher disease and PD with non-inhibitory chaperone of GCase (NCGC607) [47], (ii) mouse embryonic fibroblasts (MEFs) with Cerezyme [41], (iii) haNCSC derived dopaminergic neurons from carriers of heterozygous GBA1 mutation [44] and Gba1 transgenic mice [89] with Ambroxol. These treatments led to restored α-synuclein levels [44, 89] and/or reduced glycolipid storage [41, 47].
Even though the detailed mechanism by which deficiency of GCase activity induces accumulation of pathogenic/aggregated α-synuclein is not yet fully understood, these results suggest that the combination of i) increased levels of α-synuclein due to lysosomal dysfunction and ii) the presence of accumulated GluCer is potentially responsible for the conversion of soluble protein into aggregated/pathogenic species associated with GCase deficiency in cell cultures and in vivo.
These results are in agreement with the observation of higher levels of α-synuclein oligomers when recombinant protein was incubated with synthetic membrane systems made from GluCer in vitro.
Moreover, the levels of other sphingolipids including lactosylceramide, GM2, and GD3 were also observed in the brain of 4L/PS-NA mice (see Table notes for details) where the accumulation of GluCer and α-synuclein was also reported [40] (see Table 2). Finally, elevated levels of GM3 were reported in the plasma of PD patients [90].
Taken together, these results suggest that disruption of sphingolipid metabolism can be associated with PD pathology. Moreover, increased levels of sphingolipids can lead to increased levels of total and aggregated α-synuclein in cell cultures and in vivo.
Cholesterol
Changes in the levels of both cholesterol and cholesteryl esters were found to be associated with PD [41, 43]. Indeed, GCase deficiency not only led to higher levels of GluCer and GluSph but also to higher levels and accumulation of cholesterol and cholesteryl ester [41]. Accumulation of cholesterol was observed in skin fibroblasts [91, 92] and heterozygotic Gba1+/-MEF, Gba1 KO MEF, GBA KD SH-SY5Y cells and in CBE treated mouse primary cortical cells [41]. In addition, Gba1 KO led to an increase in the levels of both cholesterol and cholesteryl ester (15:1, 22:1, 24:1) in MEF [41]. Changes in cholesterol levels can affect lysosomal activity; in particular, increased levels were found to impair the maturation of late endosomes into lysosomes [93–95]. As mentioned in the previous section (see Sphingolipids), disruption of lysosomal activity can lead to ineffective clearance of α-synuclein and an increase in its levels [41]. These results therefore suggest that accumulation of cholesterol due to a decrease in GCase activity may indirectly increase the levels of α-synuclein by contributing to a decrease in lysosomal maturation and compromising lysosomal activity.
The relationship between high cholesterol levels and PD is, however, controversial as elevated serum levels of cholesterol were related to either an increase [96, 97] or a decrease of the risk of PD [98] or not to be associated with PD [99, 100]. These observations suggest that changes in cholesterol levels might indirectly be related to PD and changes in cholesterol derivatives, such as oxysterol and oxidative cholesterol metabolites, might better correlate with onset of PD [43, 101]. Indeed, elevated levels of oxidised cholesterol metabolites were observed in the brain of patients with Lewy body diseases and were found, unlike cholesterol, to accelerate α-synucleinaggregation in vitro and in dopaminergic cell lines [43]. In addition, increased levels of 27-hydroxycholesterol (27-OHC) were found in the cortex of patients with PD [102] and treatment of SH-SY5Y cells with this oxysterol led to an increase in the levels of both soluble and insoluble α-synuclein [88].
Oxysterols are oxidised derivatives of cholesterol which are involved in cholesterol homeostasis [103]. The main two oxysterols are 24-hydroxysterol (24-OHC) and 27-OHC and unlike cholesterol, they can cross the blood-brain barrier [104]. Under physiological conditions, the 27-OHC:24-OHC ratio is well defined [101] but under oxidative stress, conditions which has been associated with PD [105], and hypercholesterolemia, the levels of 27-OHC increase and this increase affects the 27-OHC:24-OHC ratio [106, 107]. The increase in 27-OHC was proposed to indirectly influence the levels of α-synuclein [88, 108]. In particular, 27-OHC was proposed to bind to and activate liver X receptors (LXR) which binds to the LXR response element in the α-synuclein promotor, leading to the overexpression of α-synuclein[88, 109].
In summary, these results suggest that the disruption of cholesterol homeostasis can lead to the accumulation of α-synuclein both in vivo and in cell cultures, not likely via a direct interaction between the lipid and the protein, but more probably via the disruption of other cellular processes such as lysosomal activity and activation of transcription factors, e.g., LXR, leading to increased levels of α-synuclein.
Conclusions
Interactions between α-synuclein and lipids have been reported both in vitro and in vivo to lead to the formation of a variety of assemblies ranging from amyloid fibrils, oligomeric species (toxic and non-toxic to cells) and mixed protein-lipid particles. Mainly three classes of lipids were found to be associated with pathological interactions with α-synuclein, i.e., fatty acids, sterols and sphingolipids.
It is, however, not always clear whether or not changes in lipid levels or chemical properties induce the aggregation of α-synuclein via a direct interaction with the protein or via the influence on other cellular processes which in turn lead to an increase in α-synuclein levels. Measurements of the kinetics of amyloid formation in vitro in the presence of different lipid systems allow the convenient study of direct effects of these lipid systems on α-synuclein aggregation. In addition, by tuning the solutionconditions it is possible to assess the effect of these lipid systems on each individual step of α-synuclein aggregation process. Indeed, recent findings suggest that lipid vesicles interacting with monomeric and fibrillar states of α-synuclein are likely to influence both the initiation of the reaction of amyloid formation and the amplification of the toxic aggregates. Increasing synergy betweenin vitro and in vivo studies of the interplay between lipid properties and α-synuclein aggregation, allow the mechanistic details of the effects of lipids on α-synuclein aggregation and toxicity to be established and carry the promise of the rational design of inhibition strategies.
METHODS
Estimation of the stoichiometry from circular dichroism (CD) measurements
Lipid systems inducing a change in α-synuclein CD spectrum
It has been established previously that the average percentage of α-helical secondary structure of α-synuclein when bound to lipid bilayers is 71% which corresponds to a value of a mean residue ellipticity (MRE) value of –26.2±0.4×103 deg.cm2.dmol-1 [19]. For the present work, in cases where the binding parameters (K D and n) were not available from reported CD data, the stoichiometry was estimated using the following considerations.n is defined as the number of lipid molecules interacting with 1 molecule of α-synuclein. The stoichiometry n can thus be calculated from the fraction of protein bound (xα,bound) to a given lipid system at a given molar lipid-to-protein-ratio (L:P (mol:mol)) using Equation 1:
For a given L:P (mol:mol) ratio, xα,bound can be estimated from the percentage of helicity adopted by α-synuclein (Eq. 2) (% helicity mes) or its MRE (Equation 3) value at 222 nm (MREmes) using the following equations:
Lipid systems inducing no change in α-synuclein CD spectrum
In some studies, α-synuclein was reported not to interact with a given lipid system because its CD signal was unaffected in its presence at a given L:P molar ratio. It is possible to extract the estimated lower bound of the stoichiometry using the following assumption. The noise associated with a typical CD measurement of α-synuclein can be estimated to be of order 0.3 –0.4×103 (deg.cm2.dmol-1) [19, 28]. This value corresponds to ∼ 1.2% of protein bound setting an upper bound for xα bound. In other words, the absence of change in the CD spectrum of α-synuclein in the presence of a given lipid system at a given L:P molar ratio suggests that at most 1.2% of protein is bound to this lipid system. The lower bound of the stoichiometry can then be calculated using Equation 1 and xα bound = 0.01.
CONFLICT OF INTEREST
The author has none to declare.
Footnotes
ACKNOWLEDGMENTS
The author thanks Prof. Alexander K Buell andDr Ayse Ulusoy for helpful comments on the manuscript and the Alexander von Humboldt Foundation for support.