
Abstract
Background:
Enzyme replacement therapy (ERT) with recombinant human alglucosidase alfa (rhGAA) was approved in Europe in 2006. Nevertheless, data on the long-term outcome of infantile onset Pompe disease (IOPD) patients at school age is still limited.
Objective:
We analyzed in detail cardiac, respiratory, motor, and cognitive function of 15 German-speaking patients aged 7 and older who started ERT at a median age of 5 months.
Results:
Starting dose was 20 mg/kg biweekly in 12 patients, 20 mg/kg weekly in 2, and 40 mg/kg weekly in one patient. CRIM-status was positive in 13 patients (86.7%) and negative or unknown in one patient each (6.7%). Three patients (20%) received immunomodulation. Median age at last assessment was 9.1 (7.0–19.5) years. At last follow-up 1 patient (6.7%) had mild cardiac hypertrophy, 6 (42.9%) had cardiac arrhythmias, and 7 (46.7%) required assisted ventilation. Seven patients (46.7%) achieved the ability to walk independently and 5 (33.3%) were still ambulatory at last follow-up. Six patients (40%) were able to sit without support, while the remaining 4 (26.7%) were tetraplegic. Eleven patients underwent cognitive testing (Culture Fair Intelligence Test), while 4 were unable to meet the requirements for cognitive testing. Intelligence quotients (IQs) ranged from normal (IQ 117, 102, 96, 94) in 4 patients (36.4%) to mild developmental delay (IQ 81) in one patient (9.1%) to intellectual disability (IQ 69, 63, 61, 3x <55) in 6 patients (54.5%). White matter abnormalities were present in 10 out of 12 cerebral MRIs from 7 patients.
Conclusion:
Substantial motor, cardiac, respiratory, and cognitive deficits are frequent in IOPD long-term survivors who started ERT before 2016. The findings of this study can be valuable as comparative data when evaluating the impact of newer treatment strategies including higher enzyme dosage, immunomodulation, modified enzymes, or early start of treatment following newborn screening.
INTRODUCTION
Pompe disease is a rare metabolic myopathy caused by biallelic mutations in the acid α-glucosidase (GAA) gene, resulting in reduced activity of the lysosomal enzyme GAA with consecutive accumulation of glycogen and impaired autophagy [1]. In infantile-onset Pompe disease (IOPD) GAA activity is less than 1% and patients typically present with muscular hypotonia, creatinine kinase (CK) elevation and severe hypertrophic cardiomyopathy (HCM) during the first 6 months of life [1]. IOPD is rapidly progressive and survival among untreated patients beyond the age of 18 months is exceptional [2]. Pivotal studies in 2006 demonstrated significantly improved survival, cardiac and motor outcome in IOPD patients receiving enzyme replacement therapy (ERT), suggesting that an early start of ERT yields better results, and indicating that the response to ERT is variable [3–5]. While some patients died as early as untreated subjects despite a timely start of ERT, others achieved motor milestones similar to their healthy peers [6–8]. In 2010 a negative cross reactive immunologic material (CRIM-negative) status reflecting no production of GAA protein at all and high sustained antibody titers against the recombinant human enzyme have been identified as poor prognostic factors [9–11]. With prolonged survival, it also became evident that IOPD is a multisystemic disorder, hampering swallowing, speech, hearing, and cognitive function; furthermore motor and respiratory status often deteriorate gradually even despite an initially good response to ERT [6, 12–14].
Limited effects of ERT and identification of negative outcome predictors led to treatment modifications in recent years such as variations of ERT dosage and frequency, immunomodulation, and implementation of newborn screening in some countries [11, 15–17]. Real-world studies from different countries have reported longer follow-up data with regard to motor and respiratory function, while other investigations have analyzed specific aspects of IOPD such as cardiac and cognitive function in older children [14, 18]. However, more than 15 years after approval of ERT knowledge about the problems and symptoms of school aged IOPD patients is still poor [12, 19]. The purpose of this study was to assess comprehensively the clinical status of German-speaking IOPD patients with an age of 7 years and older. This data can provide a basis for analyzing the efficacy of different treatment strategies including newborn screening, immunomodulation, increased ERT dosage, and next generation enzymes, aiming to further improve the prognosis of IOPD.
PATIENTS AND METHODS
15 patients (6 female) born before February 2015 were enrolled by 6 German and 1 Austrian center. Inclusion criteria were a confirmed diagnosis of IOPD based on reduced GAA activity, pathogenic variants on both alleles of the GAA gene, HCM at time of diagnosis demonstrated by echocardiography, and onset of clinical symptoms before the age of 6 months. The parents of all patients and if appropriate the patients themselves gave formal written consent for participation. The study was approved by the local ethics committee of the medical faculty of the University of Giessen, Germany (AZ 85/20). Data acquisition took place from June 2020 to January 2022.
A questionnaire was designed containing items related to cardiac and ventilatory status, Cross Reactive Immuno-stained Material (CRIM) status, immunomodulation, antibody titers if available, enzyme dosage applied, motor function (on the basis of WHO motor development milestones), hearing, vision, type of school attended, feeding method and other complications related to disease or ERT.
All patients were examined by one of the authors (13 by CP and 2 by MH) at last follow-up. Motor function was assessed using the Nine-Hole Peg Test (9-HPT), the Six-Minute Walk Test (6-MWT) [20] and the Quick Motor Function Test (QMFT). The 9-HPT is a standardized quantitative assessment of fine motor function, results are given as z-scores [21]. The Quick Motor Function Test (QMFT) comprising 16 items is a tool specially designed and validated for Pompe disease. Results are expressed as percentage of the maximum achievable score (100%, reflecting normal motor function) [22]. A conversation was recorded on video and articulation was classified qualitatively (no speech - substantially reduced speech intelligibility - slightly reduced speech intelligibility - normal speech intelligibility). Health-related quality of life was assessed using the parental version of the Short-Form36-Health Survey (SF-36). The maximum achievable score of this test is 100 after transformation of the raw data. Values between 0 and 100 represent the percentage of the highest possible health-related quality of life [23]. Cognitive function was assessed using the Culture Fair Intelligence Test (CFT) 1-R for children younger than 9 years and the CFT 20-R for children 9 years or older. The CFT is a nonverbal intelligence test designed to be free of cultural bias [24, 25]. The normal range is characterized by an average score of 100 with a standard deviation (SD) of 15. An IQ between 84 and 70 indicates mild developmental delay, and a score below 70 reflects intellectual disability. Magnetic resonance imaging (MRI) data of the brain were analyzed if available. MRIs (including T1-weighted, T2-weighted and fluid-attenuated inversion recovery images) were scored by assessing white matter changes in several anatomical regions as proposed by Ebbink et al. [14].
Statistical analysis
Normal distribution was tested by the Kolomogorov-Smirnov-Test. Since many variables were not normally distributed, all continuous variables are presented as median values and range. Categorical data are presented as percentages. Ventilatory-free survival and gastrostomy-free survival were depicted as Kaplan-Meier graphs.
RESULTS
Median age at onset of symptoms was 1.0 months (range 0.0–5.0), at diagnosis 4.5 months (range 1.0–31.0), and at start of ERT 5.0 months (range 1.5–31.0). Median time interval between diagnosis and start of ERT was 0.5 months (range 0.0–4.0). Three CRIM-positive patients (20%) received immunomodulation. One patient was immunomodulated after repeated allergic reactions shortly after starting ERT, another one due to his advanced age of 5 months at diagnosis and a severely reduced general condition. A third patient underwent immunomodulation at the age of 12 years due to high sustained antibody titers. Table 1 shows demographic, genetic, and therapeutic data (ERT dosage and immunomodulation), and infusion associated allergic reactions (IAR) of each patient. At the beginning of ERT all patients had increased serum CK values (range 420–2600 U/l) and HCM (left ventricular mass index value >64 g/m2). Eleven infants (73.3%) had not achieved any motor milestone while 4 (26.6%) could turn around. Six infants (40%) were fed by naso-gastric tubes.
Synopsis of demographic, genetic and therapeutic data
*NM_000152.5 was used as a reference sequence for GAA mRNA, + = CRIM-positive; – = CRIM-negative; dosage of ERT: 1 = 20 mg/kg biweekly, 2 = 20 mg/kg weekly, 3 = 40 mg/kg weekly; Neg = negative; NA = not available; IAR, infusion associated reaction.
Follow-up
Motor development
All children gained motor skills and learned to sit independently and to crawl. Seven (46.7%) reached independent ambulation at a median age of two years (range 1.2–2.2). Two lost the ability to walk at age 3 and 7 years, respectively. Figure 1 depicts the best motor function achieved at age 2 and 4 years, and at last follow-up. Median age at last clinical examination was 9.1 years (range 7.0–19.5). Six patients (40%) were sitting without support while the remaining 4 (26.7%) had lost all motor skills and had become tetraplegic at a median age of 3.0 years (range 2.1–8.3). Table 2 summarizes the patients’ clinical outcomes. Patient 8 was tetraplegic and needed assisted ventilation from the age of 2 years on, and it was decided not to adjust further the enzyme dosage to increasing weight. He died at the age of 9.4 years, 4 months after inclusion in the study, due to unstoppable bleeding from his tracheostoma. All remaining patients are still alive at the time of manuscript preparation.
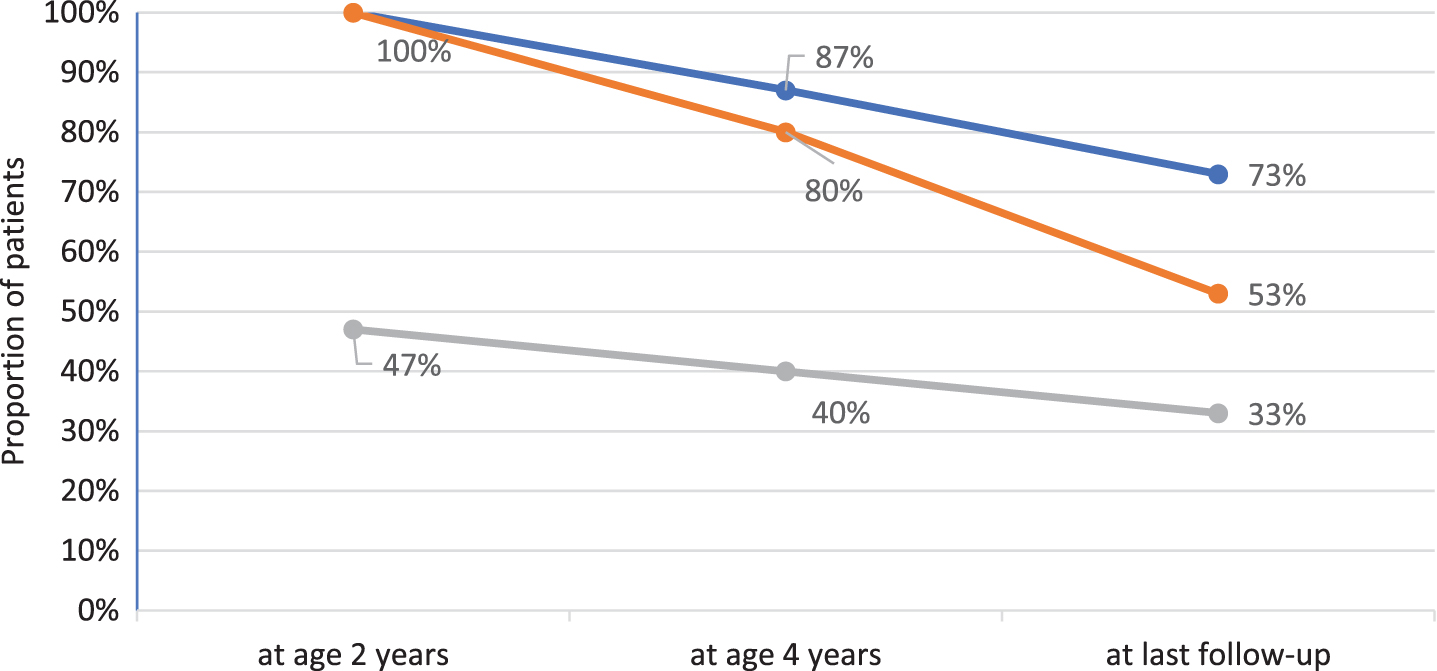
Best motor function over time in 15 IOPD patients age 7 years or older. Blue line reflects percentage of patients sitting without support, orange line mirrors percentage of patients crawling and grey line reflects percentage of ambulatory patients.
Summary of key clinical findings present at last follow-up
NA, not assessed; QMFT, Quick Motor Function Test; 9-HPT, Nine Hole Peg Test; WPW, Wolff-Parkinson-White syndrome; SVT, supraventricular tachycardia; +, i., invasive ventilation; +, n.-i., non-invasive ventilation; -, no ventilation; TF, tube feeding; *stopped test due to exhaustion.
Cardiac status
Cardiac hypertrophy normalized in all children. However, patient 8 developed again progressive biventricular hypertrophy at approximately 6 years of age. Wolff-Parkinson-White (WPW) syndrome was diagnosed in 5 patients (35.7%) and supraventricular tachycardia (SVT) was documented in another 2 (14.3%).
Ventilatory status
At last examination 8 patients (53.3%) were free of any ventilatory support, 2 (13.3%) needed non-invasive ventilation during sleep (NIV) and 5 (33.3%) were ventilated via tracheostomy. Median age at start of assisted ventilation was 5 years (range 1.7–10.8). Supplemental Figure 1 depicts ventilator-free und gastrostomy-free survival of the patient cohort. No correlation was found between age at treatment initiation and the need for ventilation or loss of ambulation.
Feeding methods/ Nutritional status
Eight children (53.3%) were solely fed orally; including one patient who was temporarily fed via a gastrostomy for 3 years. Eight patients underwent percutaneous gastrostomy at a median age of 3 years (range 9 months–12 years). Three children (20%) had a BMI below the 3rd and 3 below the 10th percentile.
Hearing, speech, vision and school performance
Hearing loss (≥40 dB) was detected in 8 out of 12 patients (66.7%). Sensorineural hearing loss was found in 5 (62.5%), combined conductive and sensorineural hearing loss in 2 (25%), and combined conductive and retro cochlear hearing loss in 1 patient (12.5%). Hearing aids were used by 4 patients. Facial muscle weakness and/or speech problems were present in all patients, 13 (86.7%) had ptosis. Speech intelligibility was not impaired in 2 patients (13.3% : patients 10, 7), slightly reduced in 6 (40% : patients 3, 4, 5, 6, 12, 15) and substantially reduced in 5 (33.3% : patients 1, 2, 9, 11, 13). The remaining 2 (13.3% : patients 8, 14) could not speak at all. Visual abnormalities were detected in 8 children (myopia in 3 patients and hyperopia in 5 with additional strabismus in 2). One patient successfully completed secondary school and is currently in his 3rd year of apprenticeship, 4 children (26.7%) attended a regular school, 3 of them had a full-time paraprofessional providing support for their physical needs, 5 children (33.3%) attended a special school for physically handicapped, one patient (6.7%) was homeschooled online, and the remaining 4 (26.7%) did not attend any school (2 were deferred from school attendance for one year due to Pompe disease and 2 were not yet send to school because of the COVID-19 pandemic).
Motor function at last follow-up
The median QMFT score was 14 (≜21.9%, range 0–73.4%) and the median z-score for the additional time required for the 9-HPT was 3.8 (range –0.12–7.4) with the dominant hand and 6.7 (range 0.7–178.5) with the non-dominant hand. Joint contractures were evident in 13 patients (86.7%). The 6-MWT was completed by 4 patients (the fifth ambulatory patient had recently suffered a tibial fracture and was not yet able to walk again for 6 minutes). The median distance walked was 262.5 m (range 200 m–373 m), the z-score based on height was –5.3 and based on age was –5.4.
Cognitive function at last follow-up
Eleven patients underwent cognitive testing. IQ scores ranged from normal development (IQ 117, 102, 96, 94) in 4 children (36.4%) to mild developmental delay (IQ 81) in one (9.1%) to intellectual disabilities (IQ 69, 63, 61, 3x <55) in 6 patients (54.5%). Three patients (20%) were unable to meet the requirements for the standardized testing and one patient (6.7%) terminated the test early due to exhaustion.
White matter abnormalities
Twelve cMRI examinations were performed in 7 patients. The patients’ ages at the time of MRI ranged from 1.1 to 9.2 years. Four patients had one, and three had two to four MRIs. MRI findings are shown in Supplemental Table 2. White matter hyperintensities were found in 10/12 MRIs from 6 patients, enlarged lateral ventricles in 3 MRIs from 2 patients, abnormalities of the basal ganglia in 2 MRIs from one patient (at age 7.3 and 8.2 years).
Quality of life
The SF-36 was completed by 14 parents (Fig. 2). Values of the sum scale physical health were below the normal mean in 13 (92.9%) patients. The largest negative deviation with –90.2 points was in the “Physical functioning” subscale, whereas the largest positive deviation was in “Mental Well-being” subscale.
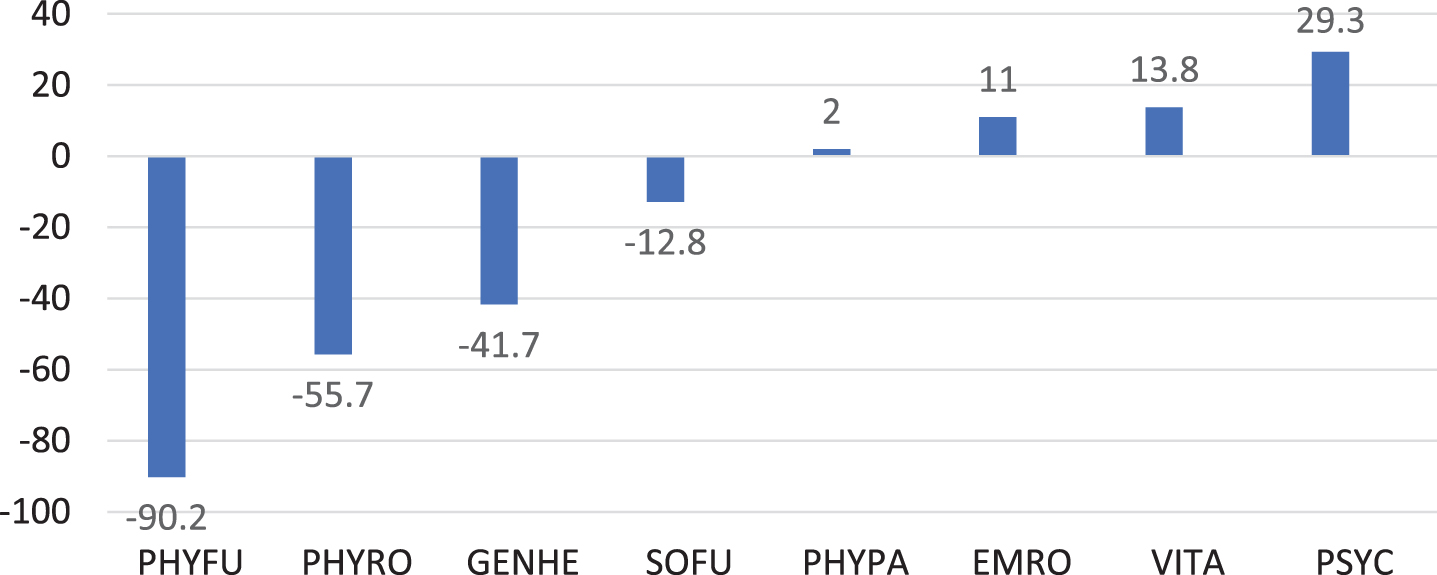
Quality of life in 14 IOPD patients 7 years or older shown as deviations from the normal mean for different domains of the SF-36. PHYFU, Physical functioning; PHYRO, Physical Role Function; GENHE, General Health Perception; SOFU, Social Functioning; PHYPA, Physical Pain; EMRO, Emotional Role Function; VITA, Vitality; PSYC, Psychological Well-Being
ERT dosing regimen
To determine the impact of different dosing regimens on motor and respiratory outcome, we compared patients who remained on the recommended standard dose during the whole observational period (20 mg/kg eow, n = 4) with subjects who received from the outset or were switched to an intermediate dose (20 mg/kg ew or 40 mg/kg eow, n = 4), and individuals who were treated from the start or were changed to a high dose (40 mg/kg ew, n = 7) (Supplemental Table 3). This showed that 3 patients who continued to receive the standard dose lost previously acquired motor milestones and required ventilatory support, while 2 out of 4 patients receiving an intermediate dose lost motor milestones and needed ventilatory support. By contrast, none of the 7 patients treated with a high dose lost an achieved motor milestone or had to be ventilated.
DISCUSSION
The purpose of this study was to analyze clinical status and quality of life in long-term surviving IOPD patients; i.e. in 15 subjects who started ERT before 2016. At last follow-up, approximately two thirds were non-ambulant, more than one-third had cardiac arrhythmias, and about half of the patients were ventilator dependent. In addition, about 50% had substantial speech, hearing and nutritional problems. Moreover, cognitive problems reflected by IQ values below the normal range or incapability to perform standardized testing was found in two thirds. The most important finding was the high morbidity among the long-term survivors, with the majority experiencing secondary deterioration of motor and respiratory functions after an initial improvement with ERT, which negatively affected their physical quality of life.
In this study, almost half of the patients achieved the ability to walk independently, however, 2 became wheelchair-dependent later. Overall, nearly 50% of our patients lost an achieved motor milestone during the course of disease. These results are consistent with real-world studies examining motor function in larger cohorts of younger IOPD patients [6–8]. They are also in line with a recent retrospective European multicenter cohort study with 86 patients showing that about 50% reaching an age of 18 months learned to walk, while at age 15 years and older only 30% were still ambulant. Our results are also consistent with the findings of Spiridigliozzi et al. who found that 6 out of 11 patients were ambulatory at a median age of 11.1 years. In contrast, a study by Ebbink et al. reported that only 2 out of 11 (18.2%) patients older than 7 years were able to walk at the last examination [14, 26]. This discrepancy could be explained by a higher rate of CRIM positive patients in our cohort and the one examined by Spridigliozzi et al. (100% CRIM-positive).
In the current study respiratory function deteriorated in half of the patients and one-third required invasive ventilation at last follow-up. This proportion is slightly lower than observed in a cohort of 17 IOPD patients aged 7.7–17.8 years (50%) [14], but is quite similar to the proportion of 35.7% in the cohort of 14 patients with an age between 1.4–14.5 years published by van Capelle et al. [18].
Our cohort was too small to draw precise conclusions on the effect of ERT dosage on motor and respiratory outcome. However, the observation that no patient treated with a high ERT dose (40 mg/kg ew) experienced a loss of motor milestones or needed to be ventilated is consistent with the study of Ditters et al. who found a positive effect of high dose ERT on survival and motor function when analyzing a larger cohort of IOPD patients [16].
Data about feeding methods and nutritional status in older children with IOPD are lacking. In our patients more than 50% were at least partially fed via gastrostomy, and none of the remaining patients could take solid foods without restrictions. In addition, nearly half of the patients had body weights and BMI values below the 10th, and one fifth below the 3rd percentile, demonstrating that nutrition and weight gain remain problematic at school age.
Speech was analyzed thoroughly by van Gelder and colleagues in younger IOPD patients. Four out of 11 patients could speak at least 10 words. All had impaired articulation with hypernasal resonance and reduced speech intelligibility [27]. Hearing was assessed by several authors [28–31]. In a Dutch study, almost 90 % of IOPD infants were found to have sensorineural hearing loss that did not worsen during follow-up [30], while this percentage was lower in two Taiwanese studies (31 % and 60 %) [15, 32]. In the present study, approximately 50% of school-aged IOPD patients had moderate hearing loss. This supports the assumption that hearing loss is rather non-progressive in IOPD. Speech intelligibility was significantly impaired or even completely lost in the majority of our patients, thus corroborating the findings of former studies and highlighting that communication is considerably impaired in IOPD patients at school age [27].
Cognitive problems in a patient with IOPD were first reported in 2010 [29]. While earlier studies examining small cohorts of pre-school children suggested that neurocognitive development is normal or only mildly delayed [15, 33], recent investigations assessing mental status in school-aged children found that IQs ranged from normal to intellectual disability [14, 26]. Moreover, repeated testing disclosed an unequivocal cognitive decline in some patients, whereas IQ values remained stable in others [14]. In our cohort, two thirds of patients had a below average IQ or could not be tested at all. Mental problems as observed in our cohort are in line with other studies stating a heterogeneous profile of cognitive impairment in IOPD patients in older age. The finding of white matter abnormalities in almost all of our patients undergoing MR imaging is in line with others [15, 34] documenting progressive leukencephalopathy in long-term surviving children with IOPD.
In this study, we investigated IOPD patients who started treatment prior to 2016. In almost all our patients the initial rhGAA dosage was 20 mg/kg every other week as recommended at that time. In the meantime, management has changed. The limited effects of ERT observed in many IOPD patients using the approved dosage has led to administration of higher doses of up to 40 mg/kg every week in many centers worldwide [6–8, 35–37]. This approach is supported by a recent multicenter cohort study including 116 IOPD patients, confirming that a high dosage of 40 mg/kg per week significantly improves patient survival and proportion of patients achieving walking abilities [16].
An immune tolerance induction protocol used for prophylaxis to preempt immune response in ERT naïve patients was first reported in 2009 [38] and the favorable effects of immunomodulation on survival and walking ability in an ERT naïve-setting were recently confirmed [39]. A negative CRIM status is associated with an increased likelihood of developing high sustained antibody titers (HSAT) and a poorer clinical outcome [8, 11]. In our study, CRIM status determination was positive in the majority of patients (86.7%). Immunomodulation was performed in three subjects. GAA antibody titers were not monitored on a regular basis in most of them. Overall, it can be assumed that greater knowledge about immunogenicity problems and increased experience with immunomodulation has led to an improved outcome of IOPD patients starting treatment today.
Trials on ERT efficacy in IOPD suggested that early treatment leads to better outcomes [3, 4]. Taiwan pioneered new born screening (NBS) for Pompe disease [15]. Comparing Taiwanese data with real world data from countries without NBS reveals superior results in ventilator-free survival and motor function, even when considering a specific genetic Taiwanese background [12, 15]. To date NBS in Pompe disease is performed in an increasing number of countries (Taiwan, Japan, and a growing number of US states) [40, 41] and there is hope to replicate Taiwan’s promising results in other patient populations [42].
As the long-term results of alglucosidase alfa are still not optimal, new enzyme preparations such as cipaglucosidase alfa and avalglucosidase alfa have been developed to improve receptor targeting and enzyme uptake [43–47]. In the Mini-COMET study, 16 IOPD patients who had inadequate response or motor function regression on alglucosidase alfa were switched to avalglucosidase alfa. Motor function improved or stabilized in all 10 patients receiving avalglucosidase alfa 40 mg/kg every other week [49]. The ongoing open label multicenter Baby-COMET study analyzes the efficacy, safety, pharmacokinetics and pharmacodynamics of avaglucosidase alfa in treatment-naïve IOPD patients (NCT04910776). Currently, the approval status of avalglucosidase alfa is variable. The drug has received US approval for the treatment of patients one year and older with LOPD, while it is authorized for all patients with Pompe disease (IOPD and LOPD) in Germany and Austria since 2021 [48]. In summary, the development of next-generation enzymes has the potential to increase the efficacy in treating patients with IOPD.
Our study has some limitations. The group of patients analyzed was relatively small and an ascertainment bias cannot be excluded. Although all physical examinations, as well as motor and cognitive testing, were performed by 2 single examiners at the last follow-up, some data are retrospectively collected from the medical records. In addition, beside CK no biomarkers were determined on a regular basis in our cohort. Especially measurement of urinary Glc4 (Hex4), a glycogen-derived glucose tetrasaccharide reflecting abnormal glycogen storage, which is now recommended by some authors for assessing response to ERT in IOPD could not be measured [50–52]. Muscle magnetic resonance imaging known to visualize progressive fatty replacement and atrophy is often used to monitor disease progression in LOPD, but its role in IOPD is less clear. Similar to what has been found in a study by Pichiecchio et al., muscle MRI performed in one patient from our cohort showed edematous swelling, but no other pathology [53, 54].
In summary, our study shows a relevant morbidity among the long-term IOPD survivors starting ERT before 2016. Notably, the aim of this study is not to denigrate ERT with alglucosidase alfa, but to describe the long-term outcome of the first IOPD generation receiving ERT. These patients may serve as a comparator group when analyzing the effects of new therapeutic strategies. Thanks to the experience gained with immunomodulation, higher enzyme doses and the development of next-generation enzymes, it can be assumed that the outcome of IOPD patients receiving ERT with recombinant GAA now and in the future will be better.
Footnotes
ACKNOWLEDGMENTS
We thank all the patients and their parents for their time and patience when participating in this study.
CONFLICT OF INTEREST
AH received honorariums for consulting activities from Sanofi-Aventis, Amicus, and Avrobio, and grants for research projects from Sanofi-Aventis. MH received unrestricted research grants from Nutricia Metabolics, SOBI, and Sanofi-Aventis as well as consultancy honoraria from Aeglea, Nutricia Metabolics, Sanofi-Aventis, and Recordati. NM received honoraria and/or consulting fees from Amicus and Sanofi-Aventis. RAH received honaria and consulting fees from Sanofi-Aventis. RS, JH, MS and CP have no conflict of interest to report.
DATA DISCLOSURE STATEMENT
Study results and individual de-identified patient data will not be available in a publicly accessible repository to protect the interests of the patients and investigators.