
Abstract
Congenital myasthenic syndromes (CMS) are rare diseases caused by mutation in genes coding for proteins involved in neuromuscular junction structure and function. DPAGT1 gene mutations are a rare cause of CMS whose clinical evolution and pathophysiological mechanisms have not been clarified completely. We present the case of two twins displaying an infancy-onset predominant limb-girdle phenotype and carrying a novel DPAGT1 mutation associated with unusual histological and clinical findings. CMS can mimic paediatric and adult limb-girdle phenotype, hence neurophysiology plays a fundamental role in the differential diagnosis.
MANUSCRIPT
Congenital myasthenic syndromes (CMS) are rare diseases caused by mutation in genes coding for proteins involved in neuromuscular junction structure and function (1). To date, at least 35 genes are known to be linked to CMS (2). DPAGT1 gene mutations represent a rare cause of CMS and a few cases only have been reported in literature. The disease course and pathophysiological mechanisms have not been completely clarified yet (3, 4).
We present here a case of two twins carrying a novel DPAGT1 mutation and displaying unusual histological and clinical findings. Furthermore, we provide a literature review on this topic.
CASE REPORT
The proband is a 59-year-old woman presenting a proximal upper and lower limb weakness and reporting concomitant early infancy motor difficulties. Clinical course was stable over years, apart from transient episodic mild worsening occurring during fever or menstrual period; a very mild fluctuant dysphagia for solid foods appeared at the age of 42. The patient was admitted at our institute at the age of 52: the neurological examination revealed mild facial and tongue muscle weakness, neck flexors and arm abductors marked weakness (Medical Research Council, MRC, 2/5), and humeral, iliopsoas, lower and upper limb distal muscles moderate weakness (MRC 3/5). Electromyography showed a diffuse myopathic pattern, whereas a 3-Hz repetitive-nerve stimulation (RNS) of ulnar, accessory, and facial nerves revealed a 37%, 61% and 14% decrement of the corresponding evoked compound muscle action potential, respectively. Extensor indicis and masseter muscle single-fiber EMG (SF-EMG) showed increased jitter and blocking. Creatin kinase (CK) level was within normal range, muscle CT scan showed normal muscle trophism without any fibro-adipose substitution, and lastly brain MRI was unremarkable. The tibialis anterior muscle biopsy revealed myopathic alterations with frequent vacuoles, including basophilic and partially periodic acid Schiff (PAS) positive material (Fig. 1a–c), but no tubular aggregates (TA) were detected. Immunohistochemistry revealed desmin accumulation (Fig. 1d), however DES and MYOT genes molecular analysis resulted normal. Glycolysis enzymes activity in the muscle was unremarkable. On electron microscopy, many small round multilamellar membranous bodies were visible in numerous fibers, either arranged in rows or in groups (Fig. 1f–h). The cytoplasm surrounding them was filled with finely granular material and contained numerous small honeybee formations, as well as vesicles, lipofuscins, small deposits of glycogen particles, large myelin-like bodies, and numerous mitochondria. Some mitochondria were gigantic and contained paracrystalline inclusions. Moreover, the sarcomeres appeared disrupted due to streaming of the z line spanning the entire sarcomere in some areas of the fibers. At last follow up, cardiac and respiratory functions were normal. At last visit, the patient was able to walk independently for around 1 km and was able to walk 10 meters in 6.8 seconds. MG composite score (5) was 7, INNCB MG score (6) was 40 and MG-ADL of 2 (7). Upper and lower limb fatigability was 25 and 22 seconds, respectively (6).
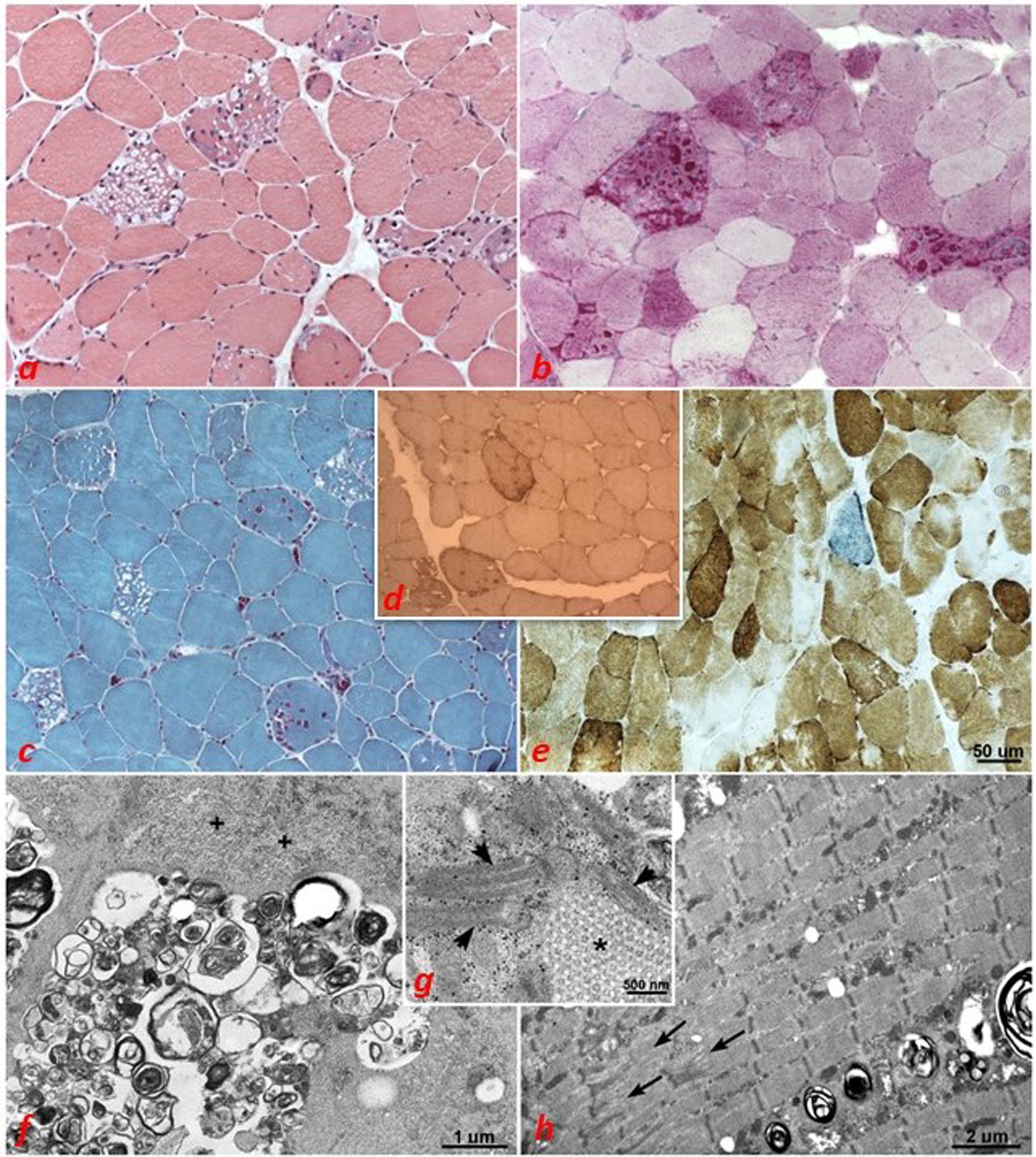
The muscle biopsy showed marked variability of fiber size, numerous central nuclei, several vacuolated fibers (a, c), with multiple vacuoles containing basophilic and partially PAS positive (b), but negative to acid phosphatase, material. Several COX negative fibers, a few fibers blue with COX/SDH staining (e), and 2 fibers with ragged red appearance were also observed (c). Vacuoles were present in both types of fibers. immunohistochemistry revealed desmin accumulation (d). On electron microscopy, many small round multilamellar membranous bodies were visible in numerous fibers, either arranged in rows or in group, cytoplasm surrounding them was filled with finely granular material (f, +) and contained numerous small honeybee formations (g, *); some mitochondria were gigantic and contained paracristalline inclusions (g, arrows). Finally, in some areas of the fibers, the sarcomeres appeared disrupted due to streaming of the z line spanning the entire sarcomere (h, arrows).
Notably, our index patient had a dizygotic twin with strikingly similar clinical features: she also presented in early infancy with walking difficulties; her neurological examination at the age of 52 revealed waddling gait, trapezius, deltoids, pectoralis major and coracobrachialis weakness (MRC 2/5), with shoulder flexion limited to 80 degrees, neck flection (MRC 3/5), finger extension and abduction (3/5) and mild ptosis. SF-EMG showed increased jitter and blocking whereas electromyography revealed a diffuse myopathic pattern. She underwent two muscles biopsies (in other centers) which reported rimmed vacuoles and type I fibers predominance. Of interest, she presented cardiac involvement with dilated cardiomyopathy in her fifth decade. A cardiac MRI demonstrated diffuse ventricular hypokinesia and severe septum and ventricles thickening resulting in reduced left ejection fraction (36%), thus she was diagnosed with class II heart failure. No cardiac perfusion defects or other systemic or genetic causes of cardiomyopathy were found. Lastly, both patients showed learning difficulties at school and required special assistance. Antibodies against acetylcholine receptor (AChR) and muscle-specific kinase (MuSK) were absent in both patients. The proband improved upper limb strength and walking distance with pyridostigmine (60 mgs, 4 times per day, started at the age of 53); further muscle strength and resistance improvement was observed when salbutamol was added (4 mgs, thrice a day at the age of 54). Conversely, her sister did not show a clear benefit with pyridostigmine (60 mgs, 4 times a day), but improved with salbutamol (4 mgs, 3 times a day, since the age of 54). At last visit proband’s sister was able to walk independently for around 500 mt and to walk 10 meters in 7.77 seconds. MG composite score (5) was 7, INNCB MG score was 200060 and MG-ADL 2 (7). Upper and lower limb fatigability was 10 and 20 seconds, respectively (6).
A custom panel for the gene involved in CMS were run on MiSeq platform (Illumina Inc., San Diego, CA). Genetic variants were annotated using ANNOVAR (http://wannovar.usc.edu/) and filtered by frequencies (1000 Genome Project, gnomAD browser and Exome Variant Server). Clinically relevant variants in the DPAGT1 gene (GenBank accession number: NM_001382.4) were found in the proband and validated by Sanger sequencing on an ABI 3130xl Genetic Analyzer (Thermo Fisher, Foster City, CA) in both patients: c.362G>A (p.R121H) and c.854A>G (p.N285S) The p.N285S variant has not been reported so far in the databases such as Leiden Open Variation Database, NextProt, Exome Variant Server and the Human Gene Mutation Database. The p.R121H variant has been reported in NextProt, and not in the other 3 databases, without any additional information. The p.R121H and p.N285S variants were classified as “likely pathogenic” and “uncertain significance”, respectively, following the ACMG guidelines. Both mutations are not reported in GnomAD exomes or other genome databases. The R121H variant is a conservative amino acid substitution, which is not likely to impact secondary protein structure as 1) these residues share similar properties, 2) this substitution occurs at a position that is conserved across species, and 3) missense variants in nearby residues (V117I, L118V, L120M) have been reported in the Human Gene Mutation Database in association with DPAGT1-related disorders (8). We submitted both detected sequence variations in the DPGAT1 gene to the Leiden Open Variation Database (LOVD: https://databases.lovd.nl/shared/genes/).
DISCUSSION
DPAGT1 encodes the dolichyl-phosphate (UDP-N-acetylglucosamine) N-acetylglucosaminephosphotransferase enzyme, which is essential to catalyse the first step in the biosynthetic pathway of the precursor oligosaccharide for N-linked glycosylation. DPAGT1 mutations have been reported as causative of CMS and other severe systemic disease with encephalopathy (DPAGT1-CDG) (9). However, whereas the pathogenic mechanism responsible for the two different phenotypes has not been established yet, it has been suggested that the primary pathogenic mechanism of DPAGT1 mutations may be the disruption of AChR-subunit glycosylation, the inefficient export of AChR to the cell surface, and consequently the reduced levels of endplate AChRs. Moreover, a missense amino acid substitution localized in exon 3 was shown to play an important role for the functioning of the neuromuscular junction (NMJ), and thus it may be causative of CMS phenotype (10). Instead, mutations in DPAGT1-CDG occur throughout the gene, although most of missense mutations are within or very proximal to a membrane-spanning section of the protein (11). In line with previous reports (12, 13, 14, 15) our two patients are compound heterozygotes for two missense mutations (p.R121H and p.N285S). The residue R121 is located in a highly conserved region of the protein, except in C. elegans where there is a lysine (K), within exon 3. The other mutation p.N285S is located in the ninth transmembrane domain of the protein, and, as demonstrated for p.Val264Met and His375Tyr mutants, both located in transmembrane domains, its substitution might be deleterious for enzyme activity. Belaya and colleagues described the first 5 patients carrying novel heterozygous mutations, with at least one mutation in exon 3 of DPAGT, presenting with a CMS phenotype: all patients had minimal cranial or bulbar involvement, but showed proximal limbs involvement. The age at onset of our patients seems to be slightly older when compared to other CMS and in all analysed cases had tubular aggregates in muscle biopsies (16). Our two patients share similar clinical and pathophysiological features of previously reported cases (Tables 1and 2). Age at symptoms onset was 5 years in our patients, which is similar to the median of 4.8 years reported in literature. They also presented with typical limb-girdle phenotype and neck flexion weakness. Notably, both our patients had intellectual disabilities, which has been reported in 4 other cases only. Furthermore, our patients had also mild bulbar symptoms: dysphagia has been previously reported in 4/14 patients. Distal weakness, another peculiar feature of our cases, was described in 8/14. Intriguingly, one of our patients developed dilated hypokinetic cardiomyopathy, which is not typically associated with DPAGT1 CMS, as only two patients have been reported to have a prolonged QT interval (14). To our knowledge, structural cardiomyopathy has not been reported in CMS related to glycosylation defects yet. As far as we know from literature, sporadic rhythm alterations have been reported in CMS patients, in particular alterations in cardiovascular autonomic functions in patients with ColQ mutations (17). Severe mitral valve insufficiency in two CMS siblings with an identical genotype has been reported, with the Authors speculating that DOK7 mutations may be associated with mitral valve disease (18). However, systemic involvement of both central nervous system and heart has been previously described as a typical feature of DPAGT1-CDG phenotype, which usually presents as arrhythmias, with no underlying structural abnormalities. Muscle biopsy seems to demonstrate heterogeneous results in this CMS subtype. Detection of TA in muscle biopsy is useful to differentiate DPAGT1-related CMS from other forms of CMS. Some reports suggest that TA accumulates over time (13, 16) and speculate that certain cellular transmembrane and secreted proteins are not appropriately glycosylated in patients with compromised function of DPAGT1, leading to their misfolding and aggregation in the sarcoplasmic reticulum with the subsequent formation of TA (16). Notably, our proband showed atypical histological findings: TA were not detected in her muscle biopsy, even if it was performed in late age, and no fibre type disproportion was noted. Conversely, vacuolated fibres containing PAS positive material and mitochondrial alterations were the most striking findings. As reported in Fig. 1, the muscle biopsy of our proband case showed numerous myopathic features. Our report highlights that it is of utmost importance to consider CMS among the differential diagnoses even in adults or in patients with suspected myopathy.
Demographic, clinical and genetic findings of previously reported cases
Laboratory, electrophysiology, and muscle biopsy findings of previously reported cases and treatment response
RNS: repetitive nerve stimulation; SFEMG: single-fibre electromyography; EMG: electromyography; CK: creatinine kinase; TA: tubular aggregates; - not known/performed or not reported. P: pyridostigmine, Sa: salbutamol; D: 3, 4-diaminopyridine.
Moreover, as highlighted in Table 1, all reported patients have decrement on RNS, when performed, and most of them also have increased jitter on SF-EMG. CMS has a highly variable clinical presentation and course in terms of severity and progression, but a favorable outcome is possible, and this condition can partially respond to therapy, so it is important to consider this diagnosis. In our cases, both patients showed relevant clinical benefit from salbutamol, which stimulates β2-adrenergic receptors improving NMJ function. Interestingly, salbutamol efficacy it has been reported in other CMS such as COLQ and DOK7 (18), but only two of the DPAGT1 mutated patients previously described in literature had benefit after therapy.
CONCLUSION
This report highlights that CMS can mimic on clinical grounds adult limb girdle phenotype and for this reason they are likely under-diagnosed. Neurophysiology plays a fundamental role in the diagnosis, but specific tests need to be performed, in particular RNS and jitter analysis, which may also be more specific than muscle biopsy. Moreover, the spectrum of CMS is not limited to muscle involvement, but it may also include CNS, cardiovascular and respiratory disorders. Expanding the genetic and clinical phenotype of this rare condition may help its recognition, which is relevant for therapeutic and prognostic purposes.
FUNDING
This work was supported/partially supported by the Italian Ministry of Health (RRC).
COMPETING INTERESTS
LM has received honoraria for speaking, advisory boards and compensation for congress participations from Sanofi Genzyme, Roche, Amicus therapeutics, Lupin and Biogen, outside the submitted work. RM has received funding for travel, meeting attendance or Advisory Board participation from Alexion, Argenx, Biomarin, Catalyst, SANOFI, Regeneron and UCB.
Footnotes
ACKNOWLEDGMENTS
LM is a member of ERN-NMD.