
Abstract
We report three siblings from a non-consanguineous family presenting with contractural limb-girdle phenotype with intrafamilial variability. Muscle MRI showed posterior thigh and quadriceps involvement with a sandwich-like sign. Whole-exome sequencing identified two compound heterozygous missense TTN variants and one heterozygous LAMA2 variant. Brain MRI performed because of concentration difficulties in one of the siblings evidenced white-matter abnormalities, subsequently found in the others. The genetic analysis was re-oriented, revealing a novel pathogenic intronic LAMA2 variant which confirmed the LAMA2-RD diagnosis. This work highlights the importance of a thorough clinical phenotyping and the importance of brain imaging, in order to orientate and interpret the genetic analysis.
INTRODUCTION
Recessive pathogenic variants of the LAMA2 gene (6q22.33; OMIM*156225), encoding the alpha-2 subunit of the heterotrimeric laminin-211 protein, cause LAMA2-related muscular dystrophies (LAMA2-RD). Laminin- 211 (or merosin) is a key component of the basement membrane of striated muscles but is also found in Schwann cells, placental trophoblasts and in the dermis-epidermis junction [1, 2].
LAMA2-RD clinical spectrum ranges from a severe form of congenital muscular dystrophy (MDC1A; OMIM 607855) [3, 4] to a milder later onset limb-girdle muscular dystrophy (LGMD R23) [5, 6]. The typical clinical features include neonatal hypotonia and delayed motor milestones in the severe congenital forms, and progressive proximal and axial weakness in the later onset LGMD forms, associated in both cases with raised CK levels. Progressive scoliosis, rigid spine, joint contractures and associated cardiac disease can also be found and raise suspicion of Emery-Dreifuss muscular dystrophy in some cases [7]. In the congenital forms feeding difficulties and respiratory involvement are commonly found. White matter abnormalities on brain imaging are one of the hallmarks of the disease [3, 8–10]. Although central nervous system (CNS) involvement may be asymptomatic, epileptic seizures are observed in up to 30% of patients [3, 11–13]. Peripheral neuropathy, particularly a sensorimotor demyelinating neuropathy, has also been reported [14, 15].
Although MDC1A represents one of the most frequent congenital muscular dystrophies (∼1/3 of patients with a diagnosis of CMD, with an estimated prevalence in UK and Italy of 0.6-0.7/100,000) [16, 17], later onset limb-girdle and milder forms represent a challenging diagnosis and can be easily overlooked.
We report here three adult siblings presenting with a slowly progressive LGMD phenotype with contractures and variable phenotype, illustrating the diagnostic challenge in LAMA2-RD.
CASE REPORT
The three patients were born to non-consanguineous parents of French origin. The proband, patient III.6, presented running difficulty since childhood. There was no neonatal hypotonia and motor development was normal (gait acquisition at 14-15 months), although Achilles contractures were present since the age of 15 months. From age 10–12 years, axial and proximal upper limb weakness and amyotrophy were noted. Joint contractures affecting elbows, wrists, fingers, hips and knees developed during childhood and adolescence and progressively worsened during adulthood. Proximal lower-limb weakness had been reported by the patient since the age of 25 years. Achilles’ tendon lengthening was performed at 28 years of age. At first examination at 30 years of age, bilateral scapular winging was noted as well as diffuse joint contractures and proximal upper and lower-limb weakness (3/5 and 2/5 on MRC scale, respectively). Rigid spine was also observed. Slowly progressive weakness led to the use of walking aids since the age of 46 years and loss of ambulation occurred at age 54 years. At last examination (age 63 years), besides proximal upper and lower limb weakness (2/5), distal upper and lower-limb weakness (3/5) was observed. There was no facial weakness, nor oculomotor or swallowing disturbances. Sensory examination showed no abnormalities. Respiratory and cardiac evaluations were normal.
His brother, patient III.7, presented with a similar clinical picture marked by predominant proximal lower limb weakness, progressive joint contractures and rigid spine. At last examination (age 58 years), proximal lower (2/5) and upper limb (3/5) weakness was noted along with rigid spine and prominent joint contractures affecting wrists, finger flexors, elbows, ankles, knees. Progressive weakness led to a reduced walk perimeter (200 meters). Cardiac evaluation was normal and last respiratory evaluation revealed a forced vital capacity of 72%.
Their sister, patient III.5, exhibited a milder clinical phenotype. She had a normal motor development although toe walking and Achilles contractures were noted since early childhood. The first muscular symptoms were observed at the age of 37 years with difficulty climbing stairs or lifting heavy weights. Slowly progressive axial and proximal weakness led to the use of walking aids since the age of 60 years. At last examination (62 years), axial (neck flexors 3/5), proximal upper and lower limbs (4/5) and Achilles contractures were noted. Respiratory and cardiac assessments were normal.
High CK levels were noted in all three patients: 648 (60 years) and –1575 U/L (39 years) in III.5, 399 (57 years) and –1200 U/L (37 years) in III.6 and 859 (43 years) and –2230 (31 years) U/L in III.7. EMG was performed in III.6 and revealed a myopathic pattern affecting proximal muscles. Nerve conductions were normal.
Muscle biopsy (deltoid) was performed in patients III.6 and III.7 (Fig. 1) and revealed in both cases mild dystrophic features. Immunostaining for dystrophin, α-dystroglycan, caveolin, telethonin, anoctamin and laminin α2 (80 kDa antibody) were normal (data not shown). Western-blot for dysferlin, dystrophin, calpain, α- and β- dystroglycans, α-sarcoglycan, caveolin and telethonin revealed no defects (data not shown).
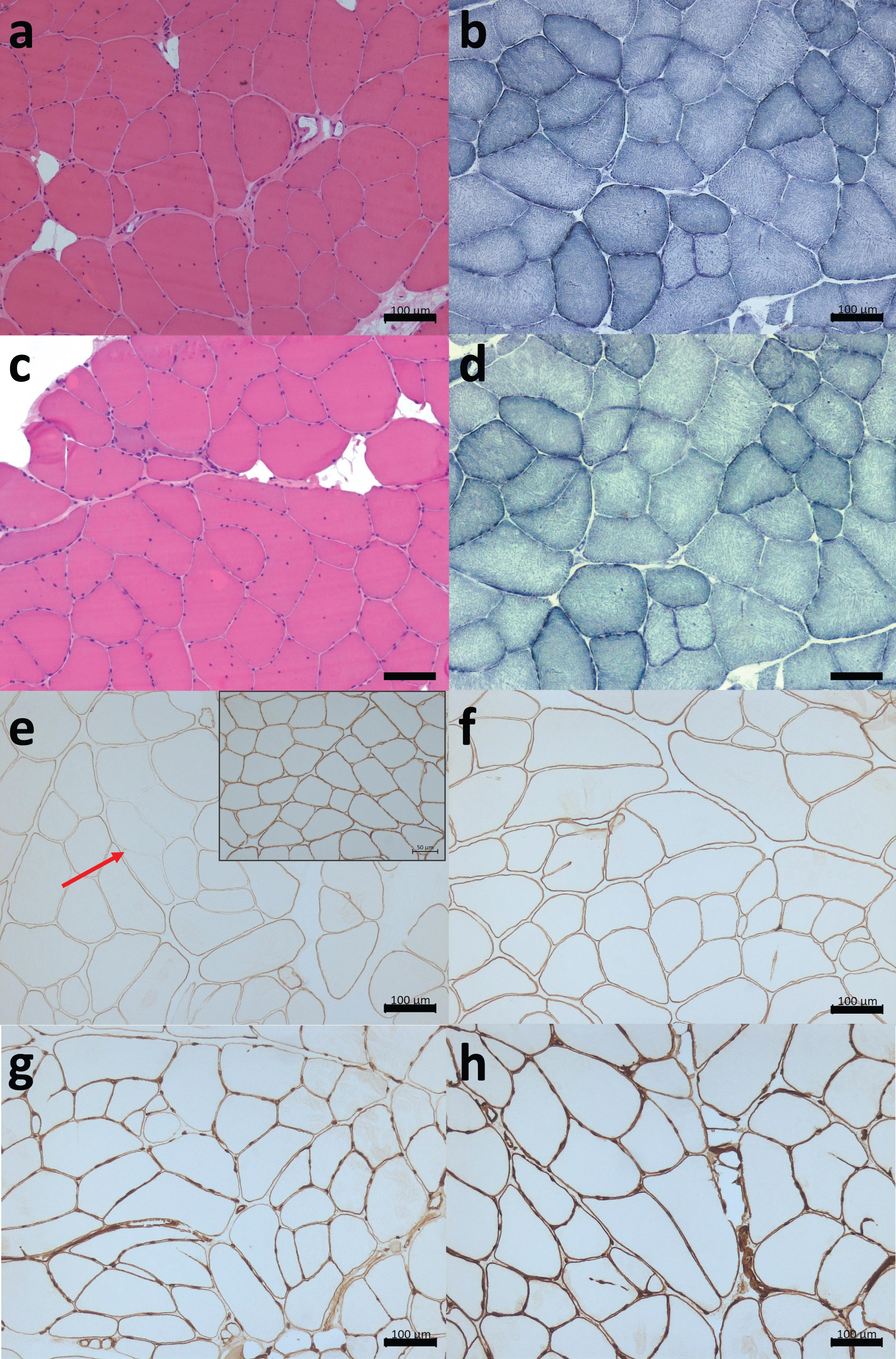
A: Skeletal muscle biopsies, frozen transversal sections. (a-b): HE staining (a) and NADH (b) from patient III.6 revealed increased endomysial tissue, fiber size variation, round and splitting fibers. (c-d): HE (c) and NADH (d) from patient III.7 showed round and splitting fibers as well as scarce necrotic fibers, mild increased endomysial tissue and internalized nuclei in certain fibers. (e-h): Immunolabelling using antibodies directed against the N-terminal part (300 kDa) of laminin-α2 (e) showing irregular labelling (red arrow) corresponding to partial deficiency and the control in the upper right corner. Weak immunolabelling with antibodies against the globular domains (80 kDa) of laminin- α2 (f). Normal labelling with laminin β (g) and laminin γ antibodies (h). HE: Hematoxylin&eosin; NADH: Nicotinamide adenine dinucleotide. Scale bars: 100 μm.
Whole-body MRI (WB-MRI) from patient III.6 revealed prominent atrophy and fatty replacement of the posterior compartment of the thigh, but also prominent involvement of the quadriceps muscles with fat substitution at their periphery contrasting with sparing of the central part (“sandwich-like sign”) (Fig. 2A). Relative preservation of sartorius and gracillis muscles was observed, along with a significant involvement of the posterior leg compartment. Given that the muscle MRI pattern was similar to that described in COL6-related dystrophy, collagen VI immunolabeling was performed on dermal fibroblast cultures from the same patient. Normal collagen VI production and accumulation in the extracellular matrix was observed (data not shown).
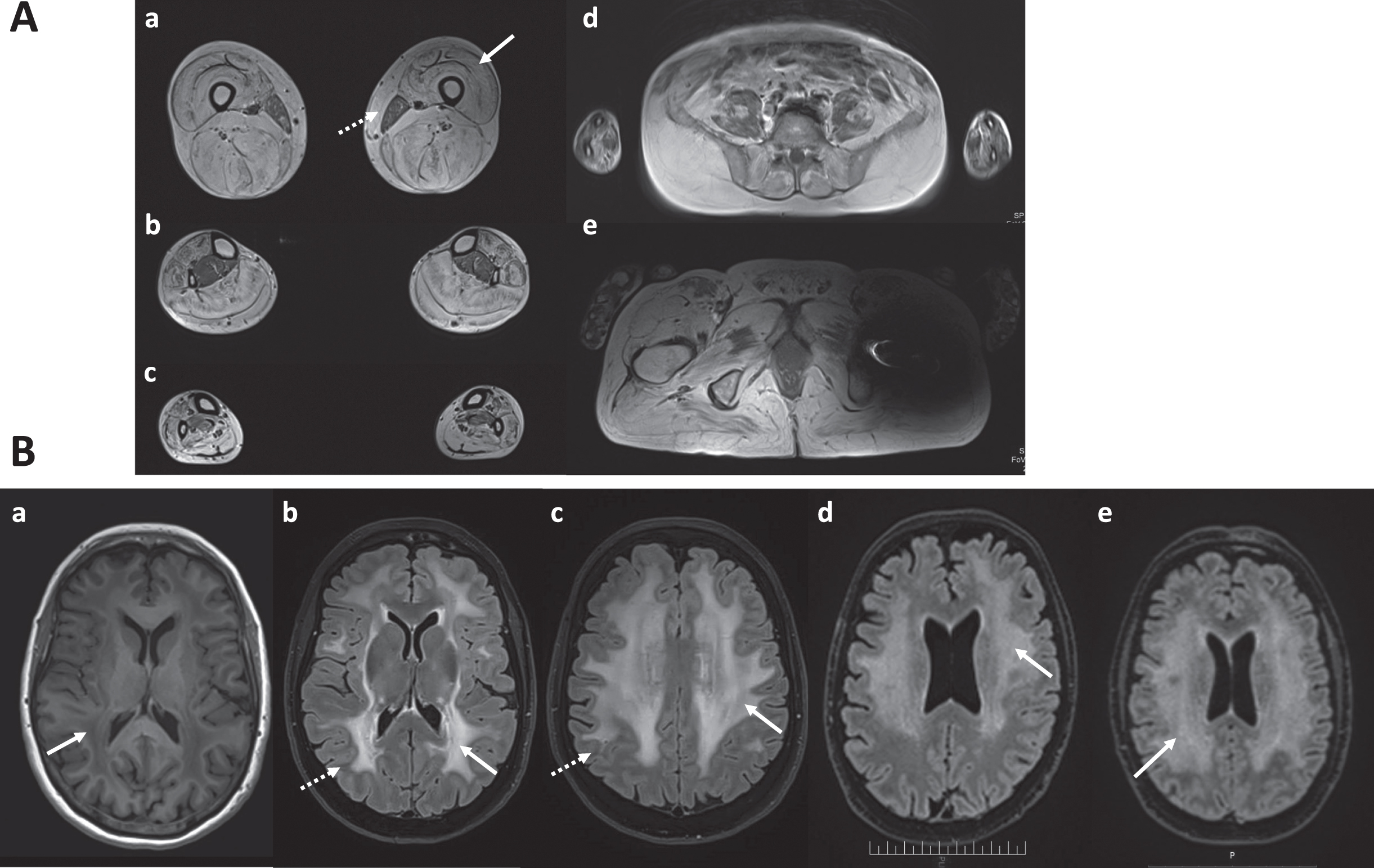
A: Muscle MRI from patient III.6. Axial T1-weighted images revealed prominent atrophy and fatty replacement of the posterior compartment of the thigh (a), as well as prominent involvement of the quadriceps muscle with fat substitution at periphery contrasting with sparing of the central part (“sandwich-like sign”, white arrow). Note relative preservation of gracillis muscles (discontinuous arrow) and significant involvement of the posterior leg compartment (b,c). (d,e): paraspinals, iliopsoas and gluteus muscles atrophy and fatty replacement. B: Brain MRI from patients III.5 (a-c), III.6 (d) and III.7 (e). (a) Axial T1-weighted image showed deep white-matter hypointensity (white arrow). (b-e): Axial T2-weighted Fluid attenuated inversion recovery (FLAIR) sequences revealed widespread symmetric supratentorial white-matter involvement in the three siblings (white arrows). Note U-fiber sparing (discontinuous arrows).
Genetic analysis (targeted Sanger sequencing) from patient III.6 excluded pathogenic variants in the DMD, DYSF, ANO5, CAPN3, LMNA, the sarcoglycan and COL6A1-A2-A3 genes. LAMA2 gene was not sequenced at the time given the mild phenotype and the initial merosin immunolabelling (80 kDa antibody), initially interpreted as normal. Whole-exome sequencing (WES) analysis was performed on DNA samples from patients III.6 and III.7, their mother and a paternal aunt (II.1). WES analysis with a recessive scenario revealed two missense variants in the TTN gene (NM_001267550): c.25104C>A, p.(Asp8368Glu), (also present in II.1) and c.14302G>A, p.(Gly4768Ser) inherited from the mother. Both TTN variants were located in the I band of the titin protein. The c.14302G>A, p.(Gly4768Ser) variant has an allelic frequency of 17/280078 in GnomAD, bioinformatics tools predicted it as possibly pathogenic (CADD, MPA, https://fraternalilab.kcl.ac.uk/TITINdb/search_page) and Clinvar and LOVD Leiden databases reported it as a variant of unknown significance (never reported in HGMD Pro). The c.25104C>A, p.(Asp8368Glu) variant has an allelic frequency of 8/243092 (GnomAD), bioinformatics tools prediction are discordant (CADD, MPA, https://fraternalilab.kcl.ac.uk/TITINdb/search_page) and Clinvar, LOVD Leiden and HGMD Pro never reported it. Several years later, the mildest patient (III.5) was seen in our center at the age of 59 years because of headache and concentration difficulties. Neuropsychological assessment revealed mild anterograde episodic memory impairment mainly due to a storage deficit. A brain MRI revealed widespread bilateral and symmetric white-matter involvement (hyperintensity on T2-weighted images and hypointensity on T1) respecting the U-fibers (Fig. 2B). A complete metabolic workup was then performed, including vitamin B1, B12, folate, homocysteine, cholestanol, cholesterol arylsufatase A and B and long chain fatty acids, revealing no abnormalities. A brain MRI was then performed for patients III.6 and III.7 also revealing extensive cerebral white matter abnormalities. No structural brain abnormalities were found in any of the siblings. These results led to reconsider the initial molecular diagnosis and raised suspicion of LAMA2-RD given the association of a limb-girdle phenotype associated with white-matter abnormalities. Following brain imaging, further genetic investigations on patient III.5 including a next generation sequencing (NGS) panel including LAMA2 gene (NM_000426.3) enabled the identification of a known pathogenic LAMA2 variant in exon 49 (rs398123383, c.6955C>T, p.(Arg2319*)) and a second intronic variant (c.8989-15T>G), classified as variant of unknown significance (VUS) predicted to alter exon 64 splicing. Both variants were subsequently found in the siblings by targeted sanger sequencing. The first variant c.6955C>T has no allelic frequency and is reported as pathogenic in LOVD and Clinvar. This variant was carried by the healthy mother (II.3) while the intronic variant was presumably paternally inherited but not carried by the parental aunt. This intronic variant has an allelic frequency of 1/250610 (GnomAD) and has never been reported in genomic databases (LOVD, Clinvar). RNA expression analysis was performed on frozen muscle samples from patient III.6 to elucidate the effect of the intronic variant. It was shown to create of a cryptic acceptor splicing site, causing a retention of the 14 last nucleotides of intron 63, leading to a frameshift with a premature stop codon 6 amino acids downstream: p.Leu2997fs*6 (Fig. 3). None of the two alleles were subjected to nonsense-mediated decay. It is worth noting that the initial immunostaining analysis of laminin α2 was performed using only the antibody directed against the 80-kDa carboxyl-terminus and revealed only a very subtle irregular staining and was initially interpreted as normal. Subsequent analysis on frozen muscle from patient III.7, performed after the discovery of the white-matter abnormalities, revealed weak immunolabelling and partial deficiency of laminin- α2 when the antibody directed against the N-terminal part (300 kDa) was used (Fig. 2e).
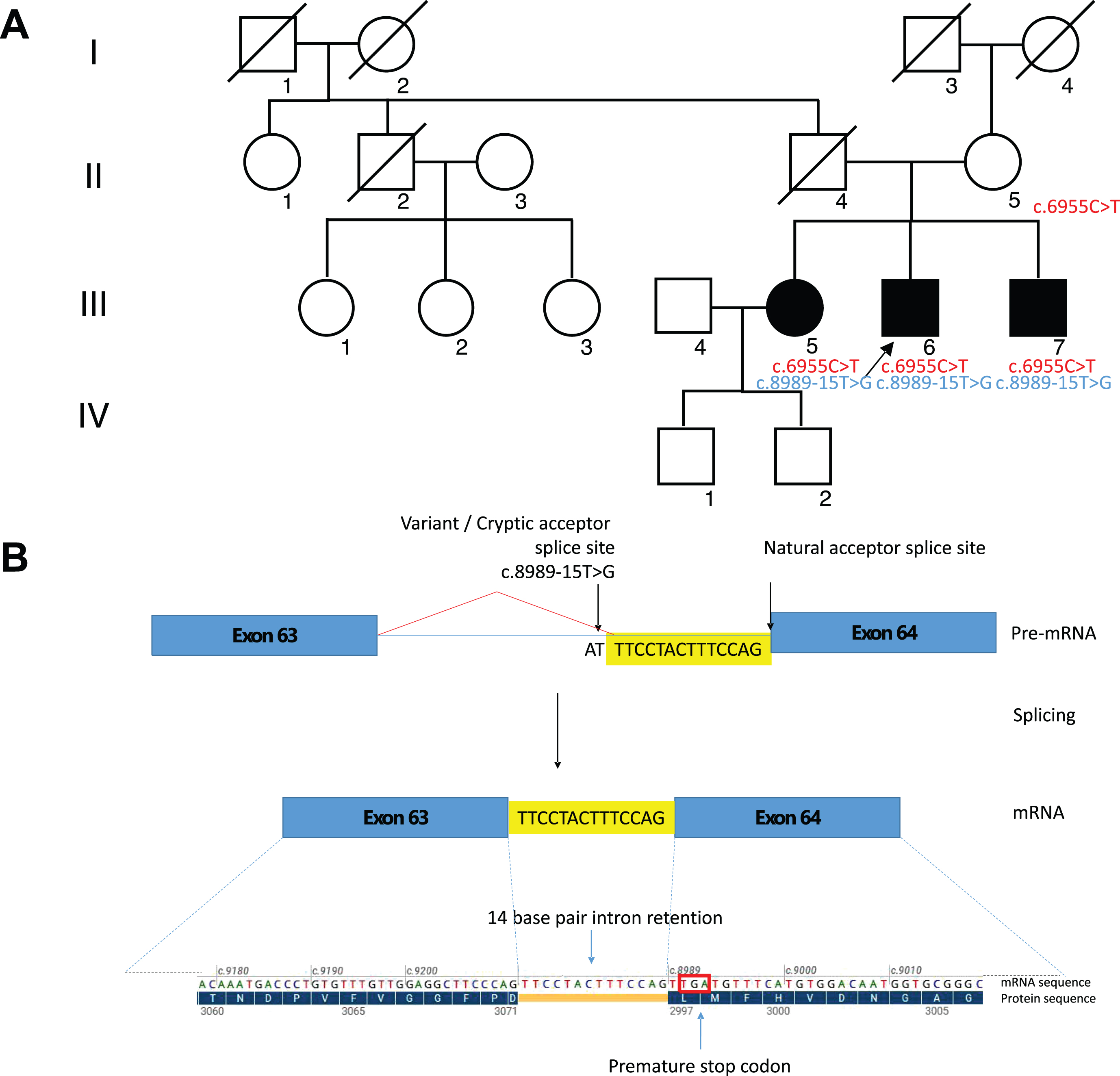
A: Family pedigree. Black-filled symbols: affected individuals. Black-arrows: index cases. The carrier status regarding the LAMA2 variants is indicated: the healthy mother carried the c.6955C>T. The parental aunt (II.1) was tested but she did not carry the intronic variant. B: Predicted functional impact of the c.8989-15T>G LAMA2 variant. Novel splicing acceptor site in intron 63 following c.8989-15T>G variant resulting in 14 base pair retention and premature stop codon. Blue bars = exons, yellow bars = intron 63 base pair retention, blue lines = introns; red lines = novel splicing event, red square = premature stop codon.
DISCUSSION
LAMA2-related muscular dystrophies include a wide phenotypical spectrum, from severe forms of congenital muscular dystrophy [4, 18] to milder later onset limb-girdle muscular dystrophy [5, 19]. This may be partly explained by the consequences of pathogenic variants on the amount of residual laminin-211 expression [20–22]. Significant intrafamilial variability can also be observed, indicating that other factors besides the gene mutation can modify disease expression and progression [7, 23]. We report here three siblings, two of them presenting from childhood with diffuse joint contractures and slowly progressive proximal weakness leading in one of them to loss of ambulation, and the third one (the eldest) with only Achilles tendons contractures in childhood and a milder adult-onset muscle phenotype.
The presence of diffuse joint contractures and proximal weakness can be noticed in a number of conditions such as COL6-, LMNA-, EMD-, FHL1-,RYR1-, JAG-2, TNPO3-, SEPN1- or TTN-related myopathies, as well as in congenital myasthenic syndromes or inflammatory myopathies [24].
Interestingly, WB-MRI in patient III.6 revealed severe involvement of the quadriceps muscle with a sandwich-like sign which is typically observed in COL6-related dystrophies (COL6-RD) [25]. Nonetheless, the COL6-like sandwich-like sign has also been reported in LAMA2-RD [7], which are also associated with atrophy and fatty replacement of subscapularis, gluteus minimus and medius muscles. Predominant involvement of the posterior thigh muscles (adductor magnus, biceps femoris, hamstrings) and soleus, as observed in patient III.6, allows distinguishing LAMA2-RD from COL6-RD [26]. Accordingly, collagen VI immunolabelling and genetic analysis of the COL6A1-3 genes revealed no abnormalities in our patients, thus ruling out the diagnosis of COL6-RD.
Two missense TTN variants were identified by WES analysis in our patients. However, interpretation of TTN missense variants remains particularly tricky due to (1) the large size of TTN gene, which has so far limited its analysis and (2) the presence of alternative transcripts that can compensate for the effects of the variants [27]. TTN-related myopathies are associated with a strikingly large phenotypical spectrum [28] but can also present since birth or during early-childhood with prominent joint contractures, rigid spine and slowly-progressive axial and proximal weakness with cardiomyopathy and a variable degree of respiratory involvement [29, 30]. Nonetheless, our patients did not exhibit cardiac involvement, which is often found in TTN-related myopathies. Interestingly, there is also increasing evidence of cardiac involvement associated with LAMA2-RD, particularly left ventricular systolic dysfunction and arrhythmia, which can be present in up to 40% of patients[31–34].
The muscle imaging was neither typical of TTN-related myopathies and brain imaging argues against the causality of the two TTN variants, as this gene has never been associated to CNS involvement and particularly with white matter abnormalities. Regarding histopathological features, TTN-related myopathies are typically associated with core lesions or other myopathic findings such as fiber size variation, internalized nuclei, cap-like lesions or whorled fibers, while muscle biopsies from our patients revealed fiber size variation, and mild dystrophic findings such as mildly increased endomysial tissue and several round and splitting fibers. Moreover, bioinformatic tools predictions for one of the variants (c.25104C>A, p.(Asp8368Glu)) were discordant.
Importantly, one of the hallmarks of LAMA2-RD is the presence of white matter abnormalities on brain MRI, which are asymptomatic in most cases but can be associated with epilepsy or cognitive impairment [8, 35]. Interestingly, the degree of white matter involvement is not correlated to disease severity nor to the degree of merosin deficiency and the amount of residual merosin expression [9, 10]. The extensive white matter abnormalities associated with the limb-girdle muscle phenotype in one of our patients led to reconsider the diagnosis, raising suspicion of a LAMA2-RD instead of TTN-related myopathy. The LAMA2 variant c.6955C>T,(p. Arg2319*) has been already reported in CMD [36], whereas the second intronic variant (c.8989-15T>G) was classified as a VUS and predicted to alter splicing, which we confirmed with mRNA transcript analysis on frozen muscle samples.
Unfortunately, due to limited coverage, the WES first analysis did not detect the intronic LAMA2 variant. Furthermore, in this case, WES analysis was conducted using the paternal aunt (II.1) sample instead of the father, whose DNA was not available. Indeed, the aunt did not carry the second LAMA2 variant. Since the most probable inheritance was autosomal recessive given the family pedigree, when the WES results were analysed only the two TTN variants emerged and were considered as variants of interest. However, pauci-symptomatic diffuse white matter involvement led us to reconsider the initial molecular results and to target genetic analysis toward LAMA2 gene. Moreover, mRNA LAMA2 expression analysis was of critical importance and confirmed the intronic variant pathogenicity. In case of splicing variants, the transcript analysis on muscle biopsy is an additional value to define the pathogenic role of the variant.
This case report highlights the importance of brain imaging in the diagnostic workup of limb-girdle muscular dystrophies. It can help the correct interpretation of the genetic analysis, which supports the pathogenicity of the so-far unreported LAMA2 variants. Since WES is becoming increasingly available, it is of utmost importance to bear in mind that a thorough clinical phenotyping, including clinical history, physical examination and ancillary tests, is mandatory to make an accurate diagnosis. In the setting of a LGMD phenotype, merosin immunostaining should be performed if muscle is available, preferably using at least two distinct antibodies targeting different fragments [37], and LAMA2 sequencing should also be included in the diagnostic workup, even in the case of later-onset or mild phenotypes. Moreover, mRNA expression analysis on muscle tissue (or dermal fibroblasts cultures) is also crucial to elucidate the pathogenic role of a given variant. Close collaboration between geneticists and neurologists is essential and genetic results should always be reconsidered when novel phenotypical findings emerge or the disease progresses.
Footnotes
ACKNOWLEDGMENTS
We acknowledge the patient’s family for their participation in this study. Part of the genetic analysis were performed within the framework of the “Solve-RD - solving the unsolved rare diseases” project, funded by the European Commission, and the “Myocapture” project, a collaborative project funded by France Genomique. We would like to thank Emmanuelle Lacène who performed the immunohistochemical techniques on muscle biopsy.
FUNDING
This work received funding support from Institut National de la Santé et de la Recherche Médicale (INSERM), Sorbonne Université Faculté de Médecine, the AFM-Téléthon association, France Génomique (Myocapture Project), National Research Agency, Investment for the Future (Grant No. ANR-10-INBS-09) as well as from the European Union’s Horizon 2020 research and innovation program, Solve RD project under grant agreement Number 779257.
DECLARATIONS OF INTEREST
Dr Gisèle Bonne is an Editorial Board Member of this journal, but was not involved in the peer-review process nor had access to any information regarding its peer-review
DATA AVAILABILITY STATEMENT
Raw data are available upon request