
Abstract
Background:
Recessive pathogenic variants in LAMA2 resulting in complete or partial loss of laminin α2 protein cause congenital muscular dystrophy (LAMA2 CMD). The prevalence of LAMA2 CMD has been estimated by epidemiological studies to lie between 1.36–20 cases per million. However, prevalence estimates from epidemiological studies are vulnerable to inaccuracies owing to challenges with studying rare diseases. Population genetic databases offer an alternative method for estimating prevalence.
Objective:
We aim to use population allele frequency data for reported and predicted pathogenic variants to estimate the birth prevalence of LAMA2 CMD.
Methods:
A list of reported pathogenic LAMA2 variants was compiled from public databases, and supplemented with predicted loss of function (LoF) variants in the Genome Aggregation Database (gnomAD). gnomAD allele frequencies for 273 reported pathogenic and predicted LoF LAMA2 variants were used to calculate disease prevalence using a Bayesian methodology.
Results:
The world-wide birth prevalence of LAMA2 CMD was estimated to be 8.3 per million (95% confidence interval (CI) 6.27 –10.5 per million). The prevalence estimates for each population in gnomAD varied, ranging from 1.79 per million in East Asians (95% CI 0.63 –3.36) to 10.1 per million in Europeans (95% CI 6.74 –13.9). These estimates were generally consistent with those from epidemiological studies, where available.
Conclusions:
We provide robust world-wide and population-specific birth prevalence estimates for LAMA2 CMD, including for non-European populations in which LAMA2 CMD prevalence hadn’t been studied. This work will inform the design and prioritization of clinical trials for promising LAMA2 CMD treatments.
INTRODUCTION
The LAMA2 gene encodes the α2 chain of laminin-211, which is a large heterotrimetric complex that represents a key component of the skeletal muscle basement membrane [1]. Biallelic pathogenic mutations in LAMA2 cause recessive congenital muscular dystrophy type 1A (LAMA2 CMD, also known as MDC1A) [2]. The phenotypic spectrum of LAMA2 CMD ranges from a severe form with neonatal hypotonia or muscle weakness shortly after birth and failure to thrive, to a milder late-onset presentation of proximal muscle weakness in childhood to adulthood [2, 3]. LAMA2 spans a 260 kb genomic region with 65 exons encoding for a 390 kDa protein [4], and more than 300 disease-associated variants in this gene have been reported [3]. Loss of function variants compose the majority of reported pathogenic variants, while missense variants represent a smaller proportion [3]. These pathogenic variants are characterized by loss of or reduced laminin-α2 protein expression, where missense variants are thought to result in a milder form of disease [3]. Currently there is no cure for LAMA2 CMD, however there are several promising treatments being investigated [5–7]. This includes gene therapy strategies such as the upregulation of LAMA1 expression to compensate for reduced laminin-α2 function [8], small molecule therapies like the anti-apoptotic compound omigapil to reduce muscle cell death [9], protein substitution therapies using the laminin-111 isoform [10], and linker proteins including mini-agrin to restore muscle basement membrane stability [11].
The prevalence of LAMA2 CMD has been estimated to lie between 1.36–20 cases per million; most of these estimates are derived from the traditional epidemiological approach of dividing the number of cases by population size [12–17]. However this approach of estimating prevalence for rare diseases can be confounded by an incomplete ascertainment of cases, and or biased by the population evaluated including due to effects of founder mutations and consanguinity [18]. The availability of large population genetic databases enables allele frequency data to be used instead for estimating disease prevalence, a method which can overcome the limitations of epidemiological approaches. Previously, we developed a Bayesian methodology for estimating the prevalence of recessive diseases from population allele frequency data, and applied it to estimate the prevalence of limb girdle muscular dystrophies [18]. In this study, we apply our Bayesian method to estimate the birth prevalence of LAMA2 CMD using Genome Aggregation Database (gnomAD) allele frequency data.
MATERIALS AND METHODS
Identification of prevalence estimates from epidemiological studies
To obtain published prevalence estimates of LAMA2 CMD from epidemiological studies, publications available on PubMed were searched using keywords ‘LAMA2 CMD’, ‘merosin deficient congenital muscular dystrophy’ and ‘MDC1A’ together with ‘epidemiology’ and ‘prevalence’. A manual search of publications which cited the retrieved studies was also performed to capture any relevant publications missed by the PubMed search.
Identification of pathogenic variants
Reported disease-associated LAMA2 variants and their interpretations of clinical significance as (likely) pathogenic, (likely) benign, or uncertain significance were obtained from National Center for Biotechnology Information ClinVar database (date of access Sep-05-2020) [19], the Leiden Open Variation Database (LOVD) (date of access Sep-09-2020) [20], and the Emery Genetics Laboratory (EGL) variant database (2020 Q4 data freeze, accessed at https://www.egl-eurofins.com/emvclass). The variants were not restricted to any specific isoforms. Any variants annotated using GRCh37 were converted to GRCh38 genomic coordinates using the pyhgvs package [21]. For variants with different clinical significance interpretations in different databases, we took a conservative approach of regarding them as variants of uncertain significance (VUS); for all other variants the listed interpretation was used. From this, a list of reported 439 pathogenic variants was obtained (including both pathogenic and likely pathogenic variants). The predicted molecular consequence of these pathogenic variants (i.e. missense, stop gain, etc) was obtained from Ensembl Variant Effect Predictor (VEP) annotation [22]. To further substantiate prevalence estimates, any predicted loss of function (LoF) variants (defined as frameshift, stop gained, splice donor and splice acceptor variants) in gnomAD v2.1 or v3 not listed in the ClinVar, LOVD and EGL databases were also included in the prevalence estimate [23].
Prevalence estimation
We used our published method to estimate the birth prevalence of LAMA2 CMD [18]. In brief, this approach involves using a Bayesian framework to estimate disease prevalence from gnomAD allele frequency data, and incorporates previously ascertained allele frequency priors obtained through modelling a beta distribution of variant data from the Exome Aggregation Consortium (ExAC) database. Allele frequency data for the finalized list of LAMA2 reported pathogenic and predicted LoF variants described above was extracted from gnomAD v2.1 and v3 via API [23]. Variant sites with an insufficient number of individuals with adequate coverage in gnomAD (allele number < 10000) were removed to reduce variability in allele frequency estimates. Variants from gnomAD v2.1 were converted from GRCh37 to GRCh38 coordinates using the UCSC liftover tool [24]. For variants in both gnomAD v2.1 and v3, the allele frequency from v2.1 was used due to the larger population size. The sizes of populations within gnomAD are provided in Supplementary Table 1. We note that this method assumes that all pathogenic variants used in calculations are independent, which may not always be the case if any are on the same haplotype.
Available code and data
The scripts and data used to estimate LAMA2 CMD prevalence are available at https://github.com/leklab/LAMA2-CMD_prevalence.
RESULTS
A list of 439 reported pathogenic LAMA2 variants was compiled from ClinVar, LOVD and EGL databases. LOVD and ClinVar contributed the majority of these variants, and ~25% (98 of 439) were reported in multiple databases (Fig. 1). 120 of the 439 reported pathogenic variants were in gnomAD; this included 113 LoF, 6 missense and 1 start lost variants (Table 1 and Supplementary Table 2). The start lost variant was excluded from the analysis to focus on LoF and missense pathogenic variants, which are well established causes of LAMA2 CMD.
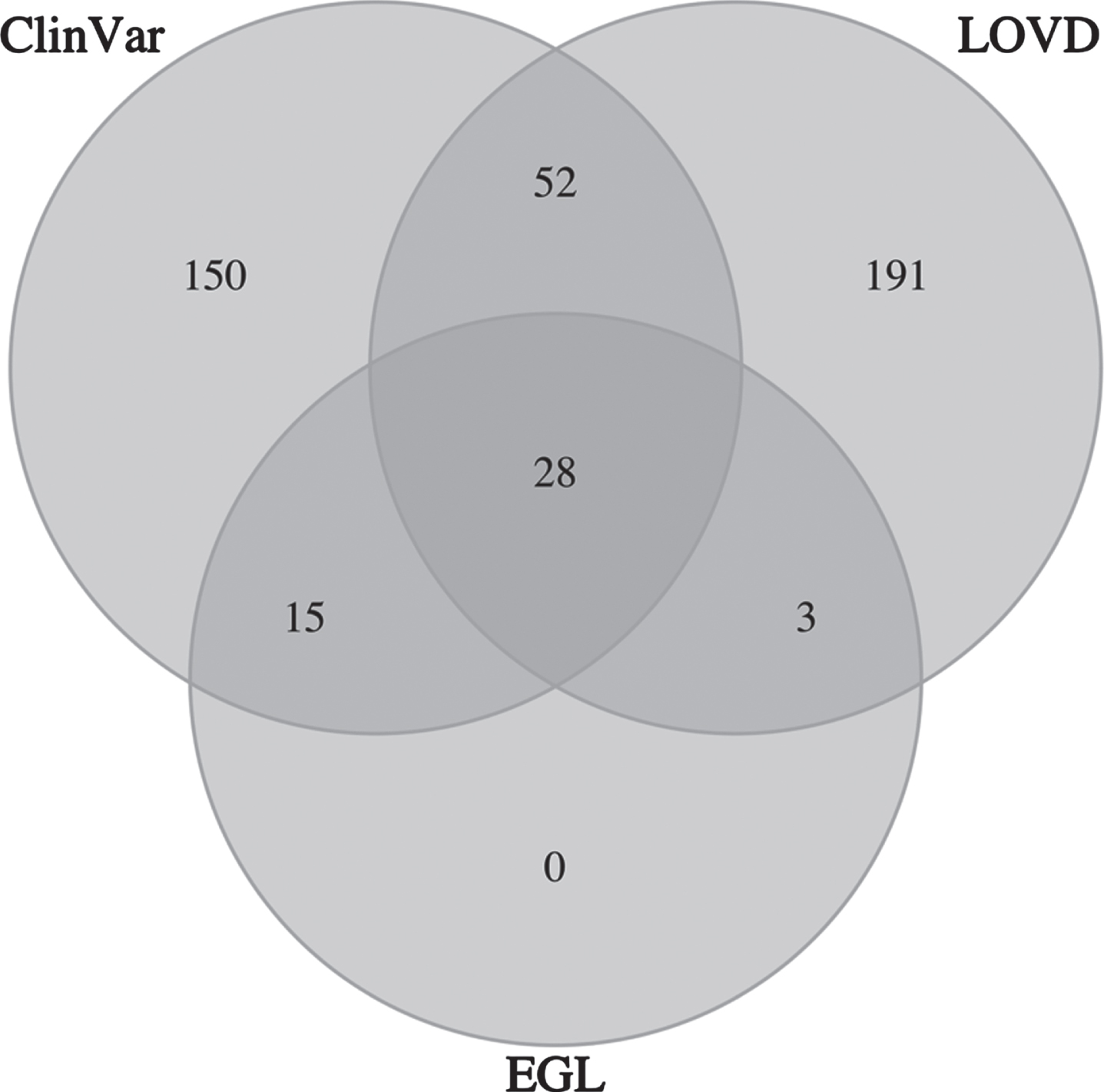
Venn diagram of LAMA2 pathogenic variants by database contribution. ClinVar is the NCBI ClinVar database, LOVD is the Leiden Open Variation Database, and EGL is the Emery Genetics Laboratory variant database. A total of 439 pathogenic variants are represented.
Counts of the reported pathogenic and predicted LoF LAMA2 variants in gnomAD that were used for the prevalence estimates. A list of these variants is provided in Supplementary Table 2
aThe reported start lost variant, and two novel LoF variants (one frameshift and one stop gain) were excluded from the prevalence estimates, as described in text.
Bayesian prevalence estimates of LAMA2 congenital muscular dystrophy based on the reported pathogenic and novel LoF variants, or only reported pathogenic variants, in LAMA2 in gnomAD
To supplement the analysis, 156 predicted LoF variants in gnomAD not in the ClinVar, LOVD and EGL databases were also retrieved (referred to as novel LoF variants, Table 1 and Supplementary Table 2). Two of these predicted LoF variants with a notably high allele frequency > 1e-4 were manually inspected and regarded as unlikely to result in loss of function. This included a stop gain variant that is rescued by a multi-nucleotide variant in the same codon (chr6:129154561A>T; ENST00000421865.3:p.(R362*) updated to p.(R362 L)), and a frameshift which duplicates the stop codon and the first base of the 3’UTR thereby preserving the stop codon (chr6:129516344 C>CTAAT; ENST00000421865.3:c.9367_*1dup). These two variants were therefore excluded from the prevalence estimates, resulting in a final list of 119 reported pathogenic and 154 novel LoF variants for the prevalence estimation (Table 1).
To assess our method of ascertaining pathogenic variants, the reported pathogenic and novel LoF variants in gnomAD (Table 1) were compared to a published list of the 14 most frequently reported LAMA2 pathogenic variants [3]. Of these 14 variants, 10 are present in gnomAD, 8 of which are included in our prevalence estimation. The two variants not represented were excluded from our study due to having conflicting variant interpretations in different databases (chr6:129280071A>C; ENST00000421865.3:p.(T821P) and chr6:129401345 G>C; ENST00000421865.3:c.5562+5G>C), highlighting our conservative approach of defining pathogenicity.
Allele frequency data for the 273 reported pathogenic and novel LoF LAMA2 variants in gnomAD was used to calculate birth prevalence estimates, including by population (Table 2). A prevalence of 8.3 per million (95% confidence interval (CI) 6.27 –10.5 per million) was estimated across all populations in gnomAD (Table 2). The prevalence estimates varied within the populations, with East Asians having the lowest estimate (1.79 per million, 95% CI 0.63 –3.36 per million) and Europeans having the highest (10.1 per million, 95% CI 6.74 –13.9 per million). Our prevalence estimates were largely consistent with epidemiological studies (Table 3), in particular for the reported worldwide prevalence, and those from England and Sweden which overlapped or approached the lower bound of the European confidence interval. In contrast, our European prevalence estimate was higher than the highest published value from Italy (Table 3). We could not compare the published prevalence for Qatar due to a lack of comparable population in gnomAD.
Summary of published LAMA2 congenital muscular dystrophy (CMD) prevalence estimates (per million) derived from traditional epidemiological methods
aNot specified. bSince 30-40% of all congenital muscular dystrophies in Europe are thought to be LAMA2 CMD [35–37], we derive this estimate by taking the 30–40% range of the reported prevalence estimate for all congenital muscular dystrophies. cThis study had genetic confirmation of LAMA2 CMD in 51 of the 81 cases. dIncluded genetic confirmation of LAMA2 CMD for all cases.
We then evaluated possible contributors to population differences in prevalence estimates. The number of reported pathogenic and novel LoF variants used in the estimates (gnomAD allele frequency > 0) in each population were broadly correlated with total population size in gnomAD (Supplementary Table 1). This suggests that the higher prevalence estimate in the European population may in part reflect their increased inclusion in gnomAD, which increases the likelihood of observing ultra-rare variation that can be used in estimation. European overrepresentation is also known for ClinVar and other databases of disease-associated variation [25]. While the effects of this on our estimates were mitigated by including all predicted LoF variants in gnomAD, the European bias of disease databases was evident for the six non-LoF pathogenic variants included (i.e. missense) given five of these were observed in European populations in gnomAD while none or only one were observed in the other populations studied (Supplementary Table 2). Differences in genetic architecture, such as founder mutations, are also likely driving disease prevalence across populations. For example, the pathogenic variant with the highest population-specific allele frequency, chr6:129250182A>AACGTGTTC; ENST00000421865.3:p.(L621Hfs*7), had a frequency > 2e-03 in the Latino/Admixed American population in gnomAD but was absent from the other populations studied (Supplementary Table 2). This frameshift mutation has been described as a ‘common pathogenic variant’ [26] and furthermore identified in multiple unrelated individuals of Portuguese or Spanish ancestry [27, 28], highlighting the contribution of population-specific variants to prevalence estimates.
We also calculated prevalence estimates using only the 119 reported pathogenic LAMA2 variants in gnomAD (Table 2). These estimates were lower across and within populations, producing a total prevalence estimate in gnomAD of 2.54 million (95% CI 1.75 –3.44 per million), showing how the inclusion of novel LoF in gnomAD increased the estimates.
DISCUSSION
Traditionally, epidemiological approaches have been used to estimate the prevalence of a disease. However, there are several issues with applying this method to rare diseases that can lower the accuracy of the results. This includes an incomplete ascertainment of cases owing to delayed or incorrect diagnosis, variation in patient ascertainment and diagnostic criteria, and or biases introduced by the population studied due to founder effects or consanguinity [18]. All these caveats are relevant for interpretation of the published prevalence estimates of LAMA2 CMD.
This study aims to address those limitations by using population allele frequencies of reported and predicted pathogenic variants to calculate birth prevalence estimates of LAMA2 CMD. The utility of the Bayesian method we applied has been previously validated using well-known recessive diseases, and other muscular dystrophies [18, 29]. Indeed, the estimates in this study were generally consistent with those derived from epidemiological studies, except for the published estimates from Italian populations which was notably below the confidence interval for LAMA2 CMD prevalence in Europeans. This may reflect regional differences in prevalence among sub-populations within Europe, and or challenges with epidemiological methods such as patient under-ascertainment.
By leveraging the availability of various populations in gnomAD, we were also able to provide estimates for non-European populations in which LAMA2 CMD prevalence hadn’t been studied previously. Our data also indicates that we may be underestimating prevalence for non-European populations that are underrepresented in databases of population and disease-associated variation, emphasizing the need to increase diversity within genetic databases. Variation in prevalence estimates across populations are likely also driven by differences in genetic architecture, including founder mutations that may be present in one population and absent from others. This was highlighted by the p.(L621Hfs*7) variant we observed with allele frequency>2e-03 in the Latino/Admixed American that was absent from all other populations studied, showing how population-specific variants can impact prevalence estimates. Capturing founder effects in underrepresented populations will thus be important for assessing regional differences in disease prevalence.
Our method for estimating prevalence has several assumptions that may impact the results [18, 29]. First, we assume that (likely) pathogenic classifications in public databases are correct, which may not always be the case. To mitigate this assumption, we did not include any pathogenic variants which had a conflicting classification in a different database. Comparison to a list of the most frequently reported pathogenic LAMA2 variants suggested that our conservative approach may lead to an underestimation of prevalence. Future studies could utilize a more permissive method for selecting pathogenic variants, such as using the interpretation agreed upon the majority of submitters, to address this. Our method for estimating disease prevalence also assumes all variants follow Hardy-Weinberg equilibrium [18]. However this can deviate due to consanguinity in populations with high inbreeding coefficients where we may observe declines in heterozygosity, as well as due to purifying selection where pathogenic variants leading to early onset forms of disease may have lower observed frequency of homozygotes relative to heterozygote carriers [30]. These are also relevant considerations for LAMA2 carrier frequency studies that use Hardy-Weinberg principles.
Another assumption made is that all predicted LoF variants in gnomAD are pathogenic. While loss of function variants in LAMA2 are a well-established cause of disease, not all predicted LoF variants in gnomAD may be pathogenic. This is evidenced by our manual curation of two predicted LoF with allele frequency>1e-4 which were revealed as likely to not be true loss of function, indicating that we may be inflating estimates by including this group of variation. Conversely, by only including missense variants reported to be pathogenic we are likely missing pathogenic missense variants from our estimates. We did not include missense variants in gnomAD due to challenges with predicting their impact on protein function, given previous studies that included missense predicted to be deleterious reported an overestimation of prevalence [18, 29]. Although missense variants are a minority of reported pathogenic variation in LAMA2, further investigation is warranted to resolve this issue.
Our approach also only incorporated single nucleotide and small insertion/deletion variants. Large structural variants which disrupt LAMA2 are an established cause of disease, representing an estimated 6% of all reported disease-associated LAMA2 variants and underlying 20% of all cases [3, 31]. The recent availability of structural variation in gnomAD opens the possibility of including this class of variation in future studies [32]. Another limitation which may lead to underestimation is that we can only incorporate pathogenic variants that are in gnomAD, which leads to ultra-rare variation not in gnomAD being excluded from prevalence estimates. The continued growth of gnomAD, and or supplementation with data from other population databases, can help to overcome this challenge.
The method applied in this study provides a framework for estimating the prevalence of other recessive congenital muscular dystrophies (CMD), given pathogenic variants in more than 30 genes have been reported to cause a recessive CMD [33]. Future studies which apply this approach to other recessive CMD genes are therefore warranted, particularly in light of promising therapeutic options under investigation for CMDs [34], although we note that the considerations described above are also relevant for the curation of variants and interpretation of estimates obtained for these genes.
In summary, we provide world-wide and population-specific birth prevalence estimates for LAMA2 CMD that are generally consistent with published results from epidemiological methods, where available. Disease prevalence information is important for the planning and prioritization of clinical trials, and therefore this work will inform ongoing and planned clinical trials for promising LAMA2 CMD treatments [5, 6]. Given our conservative method of defining pathogenic variants for inclusion, and under-ascertainment of pathogenic variants due to not including large structural variation, ultra-rare pathogenic variants not in gnomAD, and missense predicted to be pathogenic, we propose that these estimates should be regarded as a lower bound estimate of LAMA2 CMD prevalence. Future studies which address these limitations are warranted to improve our understanding of the prevalence of LAMA2 CMD.
Footnotes
ACKNOWLEDGMENTS INCLUDING SOURCES OF SUPPORT
This study was partly supported by funds received from Prothelia.
CONFLICT OF INTEREST
None.
AUTHOR CONTRIBUTIONS
J.P. and M.L. designed the study. W.L. and M.L. developed the methodology and code for the analysis. J.P. and N.J.L. performed the analyses and interpretation of results. S.A. and T.M. contributed to the interpretation of results and provided critical feedback. N.J.L, J.P. and M.L. wrote the manuscript.