
Abstract
Background:
Muscular A-type lamin-interacting protein (MLIP) has a regulatory role in myoblast differentiation and organization of myonuclear positioning in skeletal muscle. It is ubiquitously expressed but abundantly in cardiac, skeletal, and smooth muscles. Recently, two studies confirmed the causation of biallelic pathogenic variants in the MLIP gene of a novel myopathy phenotype.
Objective:
Description of the phenotypic spectrum and features of MLIP-related myopathy.
Methods:
report a patient with biallelic variants in MLIP gene with the clinical features, and histomorphological findings of MLIP-related myopathy and provide a literature review of the previously reported 12 patients.
Results:
MLIP-related myopathy is characterized by episodes of rhabdomyolysis, myalgia triggered by mild to moderate exercise, mild muscle weakness, and sometimes cardiac involvement characterized by cardiomyopathy and cardiac rhythm abnormalities.
Conclusions:
This report reviews and extends the clinical features of a novel myopathy caused by biallelic pathogenic variants in the MLIP gene.
INTRODUCTION
Rhabdomyolysis is a complicated process that destroys skeletal muscle integrity. The clinical presentation spectrum ranges from asymptomatic hyperCkemia to life-threatening conditions characterized by extreme hyperCkemia, electrolyte disturbances, acute kidney injury, and disseminated intravascular coagulation [1].
Etiologies for rhabdomyolysis are variable and include acquired (e.g., toxins or drug-induced, trauma, severe electrolytes imbalance, and inflammatory myopathies) or genetic causes. Genetic causes of rhabdomyolysis are variable and caused by different mechanisms such as a) metabolic myopathies (e.g., glycogen storage disorders, fatty acid oxidation defects & mitochondrial myopathies), b) Muscular dystrophies (e.g., Duchenne/ Becker muscular dystrophy, limb-girdle muscular dystrophies (LMGD) due to defects in FKRP, ANO5, DYSF, SGCA) and c) Defects in intramuscular calcium release and excitation-contraction coupling (e.g., defects in RYR1 and CACNA1S) genes [2, 3].
Muscle-enriched lamin-interacting protein (MLIP) is a unique gene with a specific nucleotide sequence conserved among amniotes for a minimum of seven alternatively spliced A-type lamin-interacting proteins [4]. These different splice variants are expressed differently in different tissues. MLIP is expressed ubiquitously, however, most abundantly in skeletal and smooth muscles, cardiac tissues, and brain tissue [4]. These different MLIP isoforms might have a tissue-specific function. MLIP isoforms localize beneath sarcolemma in cardiomyocytes and, therefore, play a critical role in transmitting and integrating extracellular signals in ventricular cardiomyocytes. Moreover, MLIP is found to have a role in cardiac homeostasis and cardiomyocytes’ adaptation to stress through the maintenance of balanced activity of the AMPK/AKT/mTOR pathway [5]. In addition, MLIP was found to interact with Islet 1 (Isl1), which plays an essential transcription factor for cardiac progenitor cell specification and acts by blocking agonist-induced hypertrophy of cardiomyocytes in culture [6].
MLIP role in skeletal muscle is critical. MLIP may play a role as a transcriptional co-factor that targets the regulation of genes involved in cell fate during development. It localizes to the nuclear envelope and nuclear body suggesting its interaction with chromatin specifically close to genes that encode transcription factors. Many of these genes play a critical role in skeletal muscle growth and development [7]. In addition, it plays a role in maintaining nuclear structure, chromatin organization, DNA replication, cell cycle regulation, and apoptosis [4]. Furthermore, it has a role in maintaining smooth muscle function and vascular tone [4].
Biallelic pathogenic variants in the MLIP gene have been recently described as a novel genetic cause of rhabdomyolysis [8, 9], the disease is characterized by exercise-induced myalgia, mild muscle weakness, episodes of rhabdomyolysis on the background of hyperCykemia along with cardiac involvement characterized by cardiomyopathy or cardiac rhythm abnormalities [8, 9]. Here we report a patient with a biallelic pathogenic variant in the MLIP gene causing characteristics clinical features and provide a comprehensive review of clinical and molecular phenotype in the 12 patients reported to date.
CLINICAL REPORT
Thirteen-year-old boy, born to first degree consanguineous parents with uneventful perinatal history and normal early development presented initially at 5-year old with a history of episodic muscle cramps in the legs which started at the age of 2-year. Cramps were localized to the calf muscles lasting for 5-minutes and alternated between the two legs. These episodes of cramps are usually triggered by intense or prolonged exercises or lifting heavy objects associated with muscle aches that resolved after rest for 10 minutes. The patient also described shortness of breath that usually would start after 15 minutes from initiating the exercise. He had no ocular, bulbar, proximal or distal weakness interictally. The patient had an awkward gait when he ran for a long time or played soccer for a long time. The patient denied any history of myotonia. There was no history of the second wind phenomenon. Physical examination including neurological evaluation was normal.
Investigations showed elevated creatine phosphokinase (CPK) with values fluctuating between 4000 –8000 U/L. An echocardiogram was done at the age of 7 years and a holter was done at the age of 8 years and was normal. A left quadriceps muscle biopsy performed showed mild myopathic changes on routine hematoxylin and eosin (H &E) stains. Electron microscopy (EM) revealed aggregation of glycogen granules in a moderate number of fibers either free or present within an ill-defined membrane highly suggestive of a glycogen storage disorder. A few myelin-like figures were seen. Sarcolemmal nuclei appeared normal (Fig. 1). Muscle-magnetic resonance imaging (MRI) was normal with no evidence of fat replacement. A Gene panel test to check for glycogen storage disorders and DMD gene was reported as negative. The clinical exome sequencing was initially reported as no abnormality detected. However, upon reanalysis of the exome data the patient was found to have a homozygous variant in the MLIP gene (NM_001281747.2: c.1696 C > T(p.[Gln566Ter]). Both parents were heterozygous for the same variant.
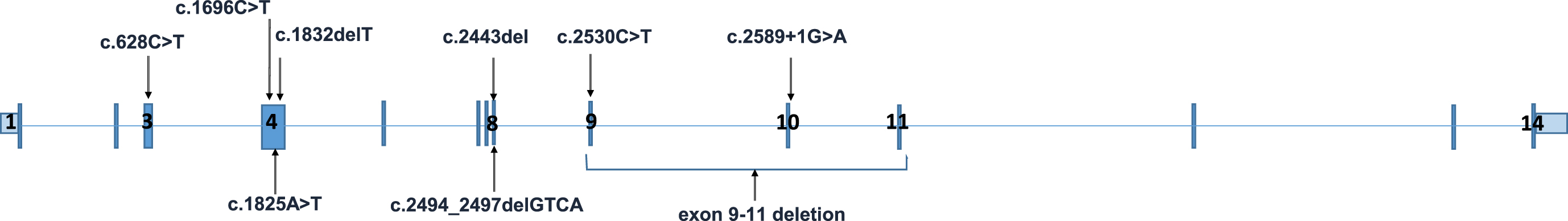
MLIP gene (NM_001281747) contains 14 exons. Variants reported to date are shown.
Family history indicated two paternal cousins with an undiagnosed muscle disease. The phenotype and clinical features were different from the proband reported in this study. Target genetic testing for the MLIP variant was normal and did not show the mutations.
DISCUSSION
Here we report a patient who presented at the age of 2 years with episodes of muscle cramps and stiffness predominantly in the calf muscles. He was reported to have muscle aches. These episodes are usually triggered by prolonged exercises or lifting heavy objects. He had no reported episodes of rhabdomyolysis. The clinical presentation of our patient is similar to what has been recently reported [8, 9]. Patients reported in Abath Neto et al. [8] and Salzer-Sheelo [9] cohorts (Table 1) were found to have mild muscle weakness, muscle ache, and susceptibility to rhabdomyolysis that are usually triggered by exercise on background elevation of creatinine phosphokinase (CPK).
Clinical characteristics of patients reported with biallelic variants in the MLIP-gene
In the study of Abath Neto and colleagues, three of the MLIP variants were localized to the large muscle-specific exon 4 and four nonsense variants localized to exons 8 & 9 (Fig. 1). These variants were speculated to either cause mislocalization of MLIP due to loss of nuclear localizing signal or result in disruption of the AT-hook-DNA binding motif. Furthermore, a homozygous frameshift variant that was found in two siblings from the same cohort was reported to localize to a highly conserved region of the MLIP. Therefore this variant was speculated to lead to the loss of the putative glycogen synthase kinase-3 (GSK3) phosphorylation site [8]. Further functional studies of RNA quantification of MLIP-predominant muscle isoforms showed a reduction in MLIP expression in affected muscles compared to controls [8].
Our patient had no episodes documented of rhabdomyolysis, and he had not reported muscle weakness. Also, he had no cardiac involvement. While three patients were reported to have cardiac involvement in Abath Neto et al. cohort (one patient with mild left ventricular dysfunction, one patient with mildly dilated aortic valve, and the third patient with patent foramen ovale) (Table 1). In addition, Salzer-Sheelo et.al reported four patients with cardiac involvement (three with cardiac arrhythmias and one patient with left ventricular hypertrophy).
The cardiac muscle involvement in this novel disease is likely explained by the fact that muscle enriched lamin interacting protein (MLIP) is expressed in different tissues but abundantly in cardiac muscles with different MLIP isoforms [4] and this protein is necessary for cardiac muscle adaptation [5]. Additionally, MLIP protein is expressed in skeletal muscles and smooth muscles [4, 7]. The skeletal muscle involvement is evident in previous reports [8, 9] in addition to our patient. The precise role of this protein in cardiac and skeletal muscles is not very well understood and necessitates further exploration. Reports showed that MLIP interacts and co-localizes with nuclear lamin A/C. It may play a role in regulating lamin A function that includes and is not limited to maintaining nuclear structure and chromatin organization [7] EM of our patient did not reveal disruption of the nuclear structure, nor was it reported by Abath Neto et al. [8]. There are similarities between laminopathies and biallelic variants in the MLIP gene including cardiac arrhythmias [9], susceptibility to rhabdomyolysis, and elevated baseline serum CPK [8]. It is noteworthy that patients with MLIP mutations present with variable clinical symptoms, some with cardiac phenotype while others with skeletal muscle phenotype or both. This needs further studies and phenotype-genotype correlation.
Light microscopic evaluation of the muscle biopsy of our patient showed non-specific myopathic changes as reported by Abath Neto et al. [8] (Table 1). However, EM showed subsarcolemmal and intermyofibrillar aggregation of glycogen granules in a moderate number of fibers. The glycogen granules were either free or present within an ill-defined membrane. There was also an occasional subsarcolemmal and intermyofibrillar myeline-like figures (Fig. 2). An occasional internal nucleus is seen. There is no evidence of intranuclear inclusions. The finding of glycogen aggregates in EM has not been reported previously [8]. The precise mechanism of glycogen aggregates is not clear. However, it might be due to the interaction of the MLIP protein with protein from pathways essential for energy production (PKM2, PDHX, DLD) involved in glycolysis and gluconeogenesis [8]. This needs further systematic exploration to establish the causal relation of metabolic derangement in all patients with MLIP-related myopathy.
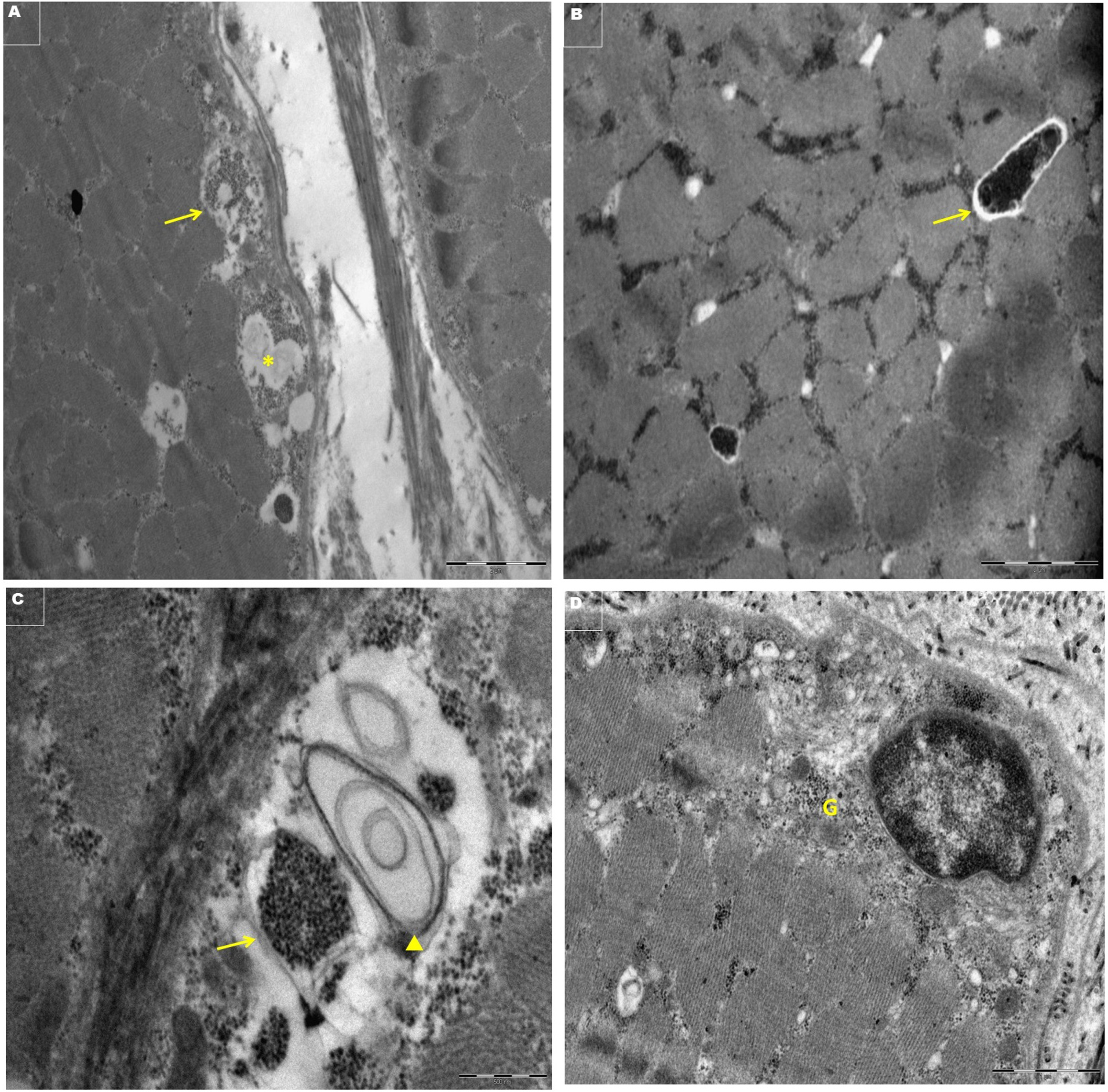
Electronmicrographs of transversely cut skeletal muscle showing. A- Subsarcolemmal non-membrane bound glycogen granules (→) and lipid (*) X9300. B- Intermyofibrillar non-membrane bound vacuole with glycogen (→) X11000. C- Subsarcolemmal glycogen within membrane bounds structure (→) and membranous myelin-like figure (▲) X30,000, D-Perinuclear glycogen (G) X 18,500.
MLIP, a nuclear lamin A/C interacting protein is distributed in the cytosol and the nucleus. MLIP is a unique gene that encodes alternatively spliced variants of unique amino acid sequences for each variant and is expressed differently in different tissues [5]. Recent reports found a tissue-specific splicing pattern. Exon 1a Mlip knock-out mice were found to significantly reduce the MLIP expression but not a complete loss. Muscle biopsies performed on the mice’s hind leg showed a large number of fibers to have centralized nuclei suggesting ongoing muscle repair [10]. Moreover, it indicates the significant role of MLIP in normal myonuclear position in skeletal muscles [11]. Furthermore, MLIP co-localizes with promyelocytic leukemia (PML) protein in the nucleus. PML bodies have been reported to play a critical role in protein post-translational modifications, transcriptional regulation, DNA damage response, and apoptosis [4, 7].
In conclusion, MLIP-related myopathy is a novel myopathy characterized by recurrent episodes of rhabdomyolysis, muscle ache, mild muscle weakness, and cardiac involvement that might manifest as cardiomyopathy or rhythm abnormalities. Further studies are required to understand the precise function of this protein and the pathogenetic mechanism in this novel myopathy, muscle biopsy findings, and the natural history of patients with this myopathy.
DATA AVAILABILITY STATEMENT
Data available on request from the authors
CONFLICT OF INTEREST
There is no conflict of interest. This manuscript has been contributed to, seen, and approved by all the authors. All the authors fulfill the authorship credit requirements. Fatema Al Amrani wrote the first draft of this manuscript. No honorarium grant or other form of payment was received for the preparation of this manuscript.
FINANCIAL DISCLOSURE
There is no financial disclosure for this project.