
Abstract
GFPT1-related congenital myasthenic syndrome (CMS) is characterized by progressive limb girdle weakness, and less prominent involvement of facial, bulbar, or respiratory muscles. While tubular aggregates in muscle biopsy are considered highly indicative in GFPT1-associated CMS, excessive glycogen storage has not been described. Here, we report on three affected siblings with limb-girdle myasthenia due to biallelic pathogenic variants in GFPT1: the previously reported missense variant c.41G > A (p.Arg14Gln) and the novel truncating variant c.1265_1268del (p.Phe422TrpfsTer26). Patients showed progressive proximal atrophic muscular weakness with respiratory involvement, and a lethal disease course in adulthood. In the diagnostic workup at that time, muscle biopsy suggested a glycogen storage disease. Initially, Pompe disease was suspected. However, enzymatic activity of acid alpha-glucosidase was normal, and gene panel analysis including 38 genes associated with limb-girdle weakness (GAA included) remained unevocative. Hence, a non-specified glycogen storage myopathy was diagnosed. A decade later, the diagnosis of GFPT1-related CMS was established by genome sequencing. Myopathological reexamination showed pronounced glycogen accumulations, that were exclusively found in denervated muscle fibers. Only single fibers showed very small tubular aggregates, identified in evaluation of serial sections. This family demonstrates how diagnostic pitfalls can be addressed by an integrative approach including broad genetic analysis and re-evaluation of clinical as well as myopathological findings.
INTRODUCTION
Limb-girdle congenital myasthenic syndrome (CMS) is a subgroup of CMS with prominent weakness of the shoulder and hip girdle muscles, while ocular and facial involvement is usually subtle [1]. One well-established cause of limb girdle CMS are biallelic variants in GFPT1 (glutamine-fructose-6-phosphate transaminase 1). This gene encodes the key enzyme in the biosynthesis of N-acetylglucosamine, an essential substrate for protein glycosylation. The clinical course of GFPT1-associated CMS (CMS12) is variable with symptom onset ranging from infancy to adulthood. However, most patients experience first symptoms in their first decade of life [2–5]. Common clinical characteristics include a slowly progressive limb muscle weakness with prominent fluctuation of symptoms and good response to esterase inhibitors [1]. The disease course is usually benign. Some of the reported patients experience gradual worsening over decades, but independent walking ability is retained in most. While weakness of the respiratory of bulbar muscles as well as ocular or facial involvement is usually absent, some patients develop a severe phenotype with respiratory insufficiency, dysphagia as well as serious facial, axial, and limb muscle weakness [2, 3, 6].
Repetitive nerve stimulation typically shows a decrement and, interestingly, needle electromyography (EMG) may demonstrate myopathic changes in proximal muscles [2–4]. In muscle biopsy, mild myopathic changes are frequently observed and, highly indicative, tubular aggregates can be identified in the majority of specimen [4].
Here, we report on a family with three affected siblings suffering from a progressive atrophic proximal muscular weakness with respiratory involvement. Based on the initial findings in muscle biopsy, a non-specified glycogen storage disease was suspected until patients’ death in adulthood. Genome sequencing finally revealed biallelic variants in GFPT1, whose pathogenicity was further confirmed in histological and biochemical analyses. This case demonstrates how diagnostic pitfalls can be overcome by the interplay of genetic analysis and re-evaluation of myopathology as well as clinical phenotyping.
CASE REPORTS
Clinical phenotypes and initial diagnostic workup
The eldest child (individual II.1) of non-consanguineous healthy parents has not been examined in our neuromuscular department. According to the sparse medical records available, there was severe early-onset muscle weakness. Motor milestones were severely delayed. The boy could neither stand nor sit independently at the age of one year. At 5 years of age, he was able to pull himself up at objects and stand with support. Walking without aids was never achieved. EMG revealed signs of a myopathy. In the documented reports of muscle biopsy, a vacuolic degeneration was described. He suffered from recurrent pneumonia. Furthermore, swallowing and eating became increasingly difficult, and he died at the age of 18 due to respiratory insufficiency.
The second son (individual II.2) first presented to our neuromuscular center at the age of 41 years for reevaluation of a progressive limb-girdle weakness. Prior to the examination in our department, there had been several assessments in other neuromuscular centers. The suspected diagnosis of the pre-treating colleagues at that time was a glycogen storage myopathy, with signs of Pompe disease. He reported slowly progressive muscle weakness since childhood. At the age of 18 months, he had been able to walk without support; however, his gait was abnormal. On clinical examination, generalized muscular atrophies, proximal muscular weakness with waddling gait, and positive Gower’s-sign as well as mild bulbar symptoms were evident (Fig. 1A). Deep tendon reflexes were absent in the upper limbs and were normal in lower extremities. Cognitive function was normal, similar to his siblings. There was no evidence of cardiac manifestation. Pulmonary function testing revealed a severe ventilatory defect. Creatine kinase (CK) levels were mildly increased (2-fold). EMG studies revealed pathologic spontaneous activity including high-frequency (myotonic and pseudo-myotonic) discharges alongside with signs of a myopathic process. Repetitive nerve stimulation was not performed. Muscle biopsy was indicative of a vacuolated myopathy with increased glycogen storage. Based on the distribution of vacuoles and glycogen deposition, in combination with enhanced acid phosphatase reactivity in some necrotic muscle fibers, glycogenosis type II (Pompe’s disease) was suspected. However, in photometric analyses, acid alpha-glucosidase enzyme activity was found to be in normal ranges. Furthermore, a gene panel analysis (38 genes) excluded pathogenic variants in the GAA gene, as well as in AGL and GBE1 (resulting in other glycogen storage diseases), whereas GFPT1 was not included [7]. Hence, a non-specified glycogen storage myopathy resembling Pompe’s disease was diagnosed, while a precise diagnosis could not be made during the patient’s lifetime. The patient died at the age of 49 years, probably due to respiratory insufficiency.
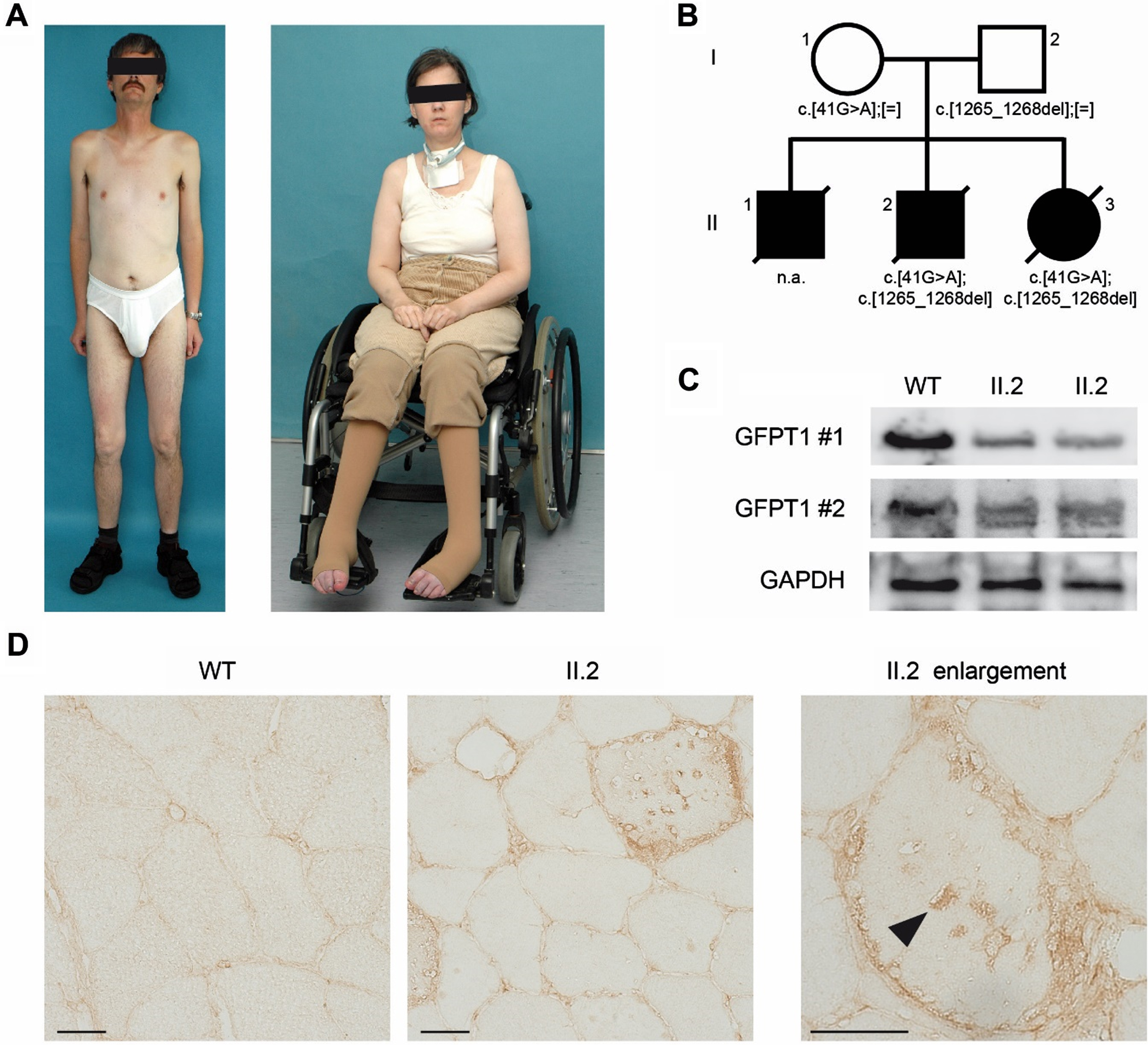
Clinical phenotype and GFPT1 protein evaluation.
The youngest female child (individual II.3) was already noticed post-partum with respiratory distress. Similar to her brother, there had been several examinations in different neuromuscular centers before we saw her, and a glycogenosis was consistently suspected. Motor development was significantly delayed: she was able to walk with personal support at the age of 18 months, with a wheeled walker at age of 3 years, and without support at 6 years of age. Since then, muscular weakness had been slowly progressive, and at the age of 15 years, she was wheelchair-bound. Neurological examination at the age of 38 revealed a proximal atrophic tetraparesis (Fig. 1A) with preserved tendon reflexes, dysphagia, and dyspnea at rest and speech. Because of recurrent pulmonary aspiration, tracheostomy had been performed. Due to progressive ventilatory defect and hypercapnia, nocturnal positive pressure ventilation had been established. EMG showed myopathic changes, while CK was normal. A muscle biopsy had been performed in an external hospital at 32 years of age. Written reports suggest a vacuolated myopathy with sarcoplasmic glycogen deposits. No tubular aggregates were reported. Thus, Pompe’s disease was suspected. However, biochemical analyses failed to confirm acid α-glucosidase deficiency. At the age of 40 years, the patient died of pneumonia after recurrent pulmonary infections.
Genome sequencing and evaluation of variant pathogenicity
Due to sustained request of the parents for diagnostic clarification and genetic counseling of the family, DNA was extracted from frozen muscle tissues of individuals II.2 and II.3 and from blood samples of both parents. No tissue from individual II.1 was available for DNA extraction. After obtaining the written consent of the parents, all available biosamples were processed using TruSeq DNA Nano (Illumina Diego, California, USA) kits. Generated libraries were sequenced as 2×100 bp paired-end reads on a NovaSeq 6000 System (Illumina Diego, California, USA). The average coverage of the four genomes ranged from 36.79X to 62.49X. Data analysis was performed with the megSAP pipeline (https://github.com/imgag/megSAP) and clinical variant prioritization was done as described previously [8].
Two variants in GFPT1 (NM_001244710.2) were identified in both affected individuals (II-2 and II-3), while parents were each heterozygous for one of the variants, confirming a compound heterozygous state in the patients (Fig. 1B). Both variants were absent in gnomAD (https://gnomad.broadinstitute.org/) and predicted to be deleterious in silico. The missense variant (c.41G > A; (p.Arg14Gln)) has been described once in the literature in association with CMS, with no further clinical information available [9]. The truncating variant (c.1265_1268del; (p.Phe422TrpfsTer26) has not been reported so far.
To further address the pathogenicity of the identified GFPT1 variants, tissue specimen and patient-derived fibroblasts of individual II.2 were analyzed with standard procedures previously described [10]. Protein extracts from primary patient-derived fibroblasts were studied by immunoblotting using two different anti-GFPT1 antibodies directed against distinct epitopes of the protein (Proteintech Group 14132-1-AP; NovusBio NBP2-52474), while GAPDH (Santa Cruz sc-47724) served as reference protein. Decreased GFPT1-protein levels were found in patients’ fibroblasts as compared to a healthy control (Fig. 1C). Furthermore, immunohistochemical staining of GFPT1 protein (using NovusBio NBP2-52474 antibody) showed a moderately reduced reactivity in the patient’s muscle biopsy sample as compared to healthy specimen (Fig. 1D). Several vacuoles appeared to contain aggregates of GFPT1 protein (Fig. 1D, arrowheads).
Reevaluation of myopathology
Muscle biopsy of individual II.2 was taken from the biceps brachii muscle at 41 years of age. Myopathic features including increased variation in fiber size, several internalized nuclei and slightly enhanced endomysial connective tissue were seen (Fig. 2A). In nicotinamide adenine dinucleotide-staining (NADH), single cells (1–2 per 100 myocytes) showed very small sarcoplasmic aggregates, that were initially classified as an unspecific perturbed mitochondrial enzyme distribution. However, reevaluation using serial sections showed a fuchsinophilic reaction of these aggregates in Gomori trichrome staining, with an intermediate reactivity of these aggregates in succinate dehydrogenase-staining (SDH), while no aggregates were present in cytochrome c oxidase-staining (COX). This indicates tubular aggregates. (Fig. 2B).
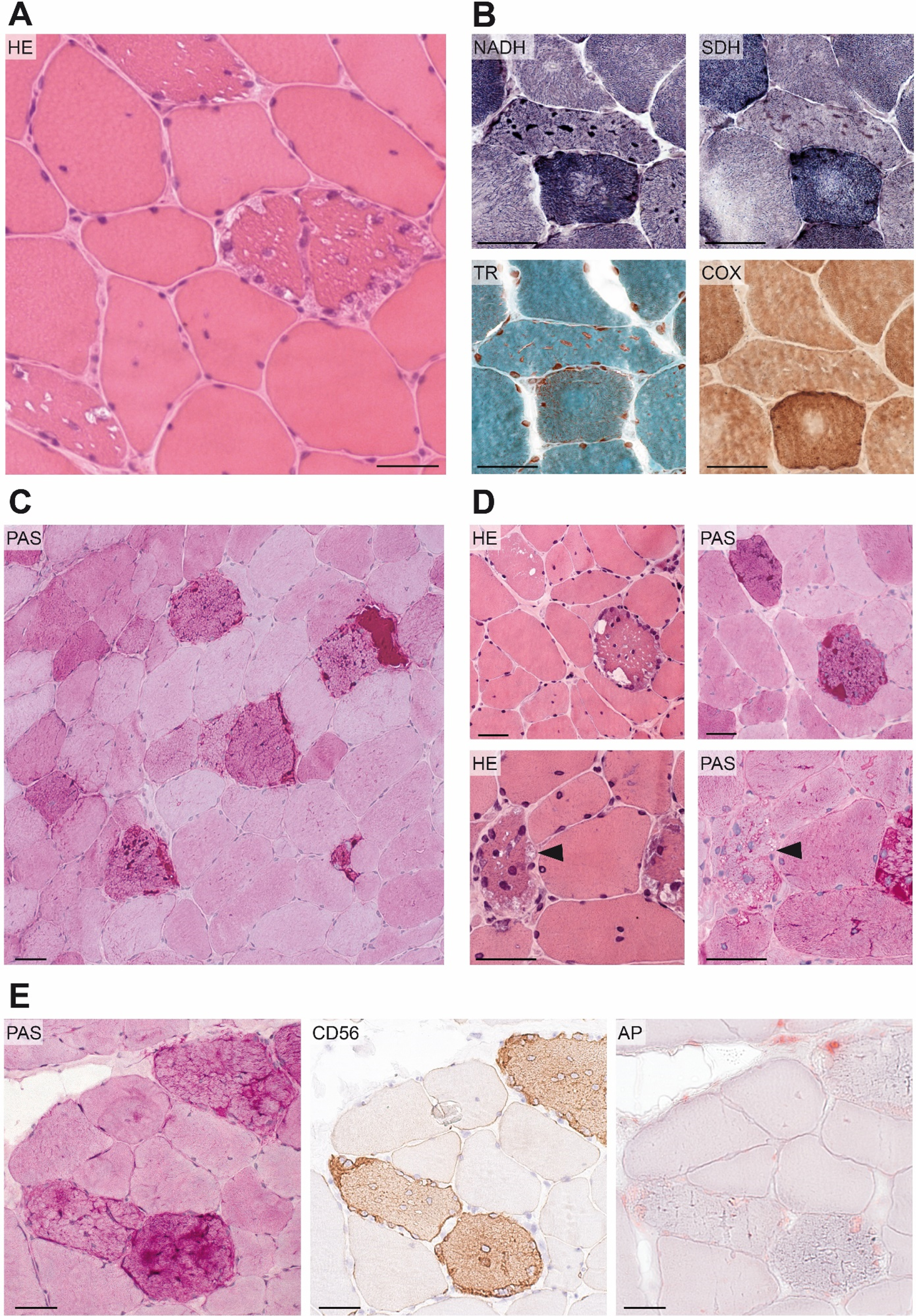
Reevaluation of myohistology
Several fibers were found to contain multiple vacuoles that appeared to be non-rimmed and empty in HE staining (Fig. 2A). While vacuoles were present in both fiber types, there was an impressive predominance in type-I-fibers. Several fibers showed marked deposition of Periodic acid–Schiff-positive (PAS-positive) material throughout the whole sarcoplasm (Fig. 2C), which was identified to be glycogen using additional diastase treatment (not shown). In most cases, vacuoles seemed to contain glycogen (Fig. 2D, upper panel). In contrast, vacuoles of some fibers appeared optically empty, and negative for PAS staining (Fig. 2D, lower panel). There was a slightly increased reactivity of acid phosphatase in few muscle fibers. However, reactivity was not localized to (subsarcolemmal) vacuoles in most cases and did not consistently colocalize with glycogen accumulations (Fig. 2E). Enhanced glycogen deposition was restricted to denervated muscle fibers, identified by immunohistochemical CD56- (NCAM-) staining (Fig. 2E).
Electron microscopy showed several optically empty vacuoles as well as autophagic vacuoles. Furthermore, there were plenty of autophagic vesicles, not associated with autophagic vacuoles and distributed throughout the whole muscle fiber (Fig. 3A and B). None of the vacuoles contained glycogen. Also, no glycogen deposits were found in the cytoplasm of the examined muscle fibers. Several nuclei showed a striking morphology with invaginations of the nuclear lamina (Fig. 3C). The area investigated by electron microscopy did not include any neuromuscular junctions or tubular aggregates. Due to the lack of additional material, it was not possible to facilitate additional investigations in terms of glycogen deposition, tubular aggregates, or motor endplates.
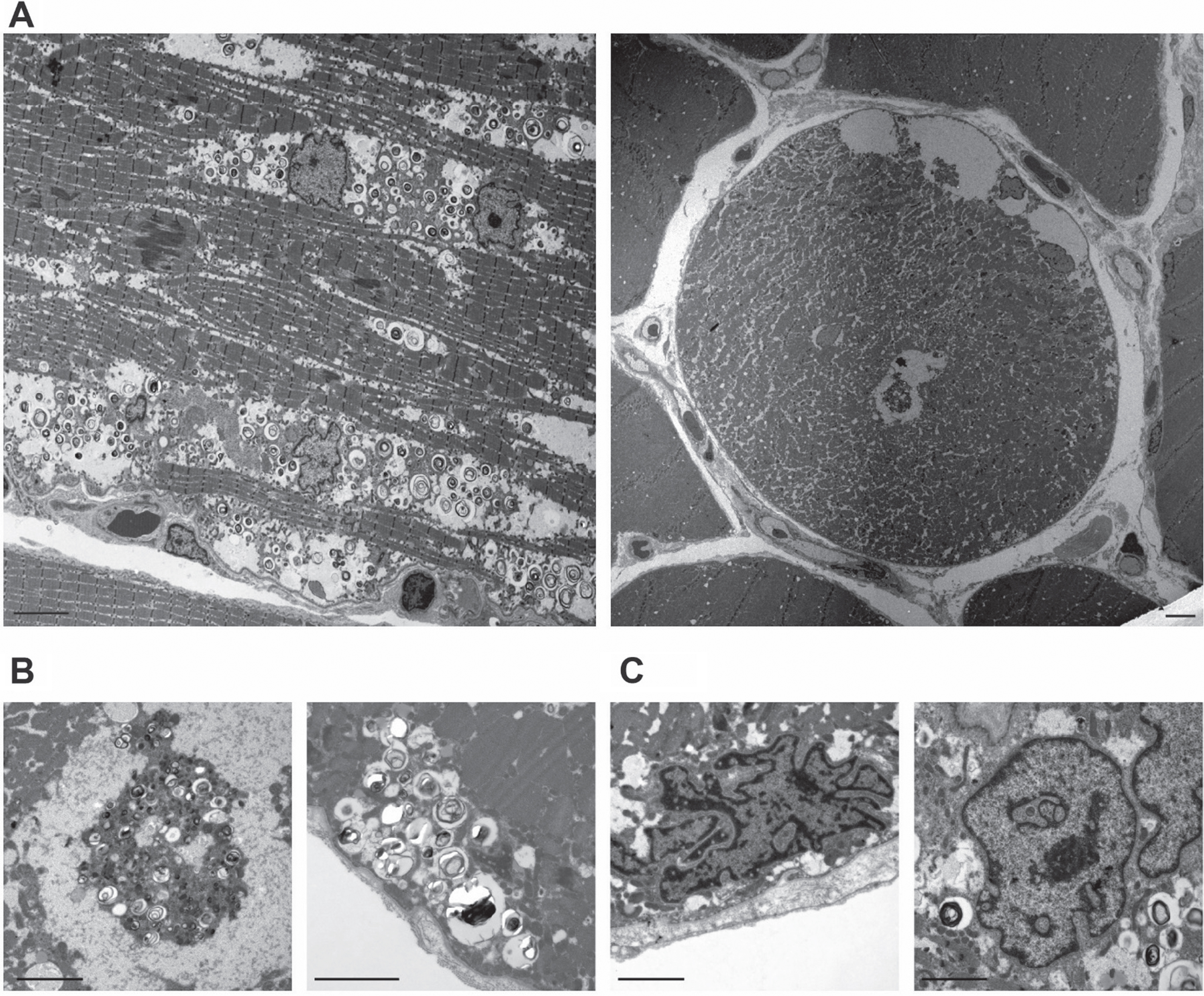
Ultrastructural analyses
DISCUSSION
We report on a family with three siblings affected by CMS12 due to two compound-heterozygous pathogenic variants in GFPT1. While the missense variant (c.41G > A; (p.Arg14Gln)) has previously has been associated with CMS, a novel truncating variant (c.1265_1268del; (p.Phe422TrpfsTer26)) was identified in our family. This variant predicts a premature stop codon at the sugar isomerase domain of GFPT1. In line with a functional relevance of the variants, GFPT1 protein levels were found reduced in muscle tissue of individual II.2, as compared to a healthy control. Furthermore, reduced GFPT1 reactivity was evident in immunohistochemical analyses of the same patient. These results are in line with a classification of the truncating variant as pathogenic (ACMG class 5) [11]. The observed accumulation of GFPT1 in sarcoplasmic vacuoles may be indicative of dysfunctional protein folding and subsequent protein aggregation. Hence, the results of this study further support the pathogenicity of the identified GFPT1 variants.
The most striking differences from previous cases were observed in myohistology. A prominent feature in this study was the excessive glycogen storage in muscle fibers, which has never been reported before in the context of CMS12. In retrospect, this myohistological phenomenon must be considered a diagnostic pitfall, as it has substantially shifted the focus of the diagnostic efforts toward glycogen storage myopathies. The aetiology of sarcoplasmic glycogen accumulation remains elusive. GFPT1 is the key enzyme of the hexosamine biosynthesis pathway (HBP), which provides the substrates for protein glycosylation [2]. However, both glycogen synthesis and the hexosamine biosynthesis pathway share glucose-6-phosphate (Glc-6-P) as primary substrate, which is either directly used for glycogen synthesis or –after conversion to fructose-6-phosphate (Frc-6-P) –is introduced into glycolysis or HBP, respectively [12]. In this regard, disruption of HBP could potentially lead to enhanced glycogen synthesis due to overabundance of Glc-6-P. However, only 2–5% of cellular Frc-6-P is used for HBP, making this hypothesis less likely. In this study, excessive glycogen accumulation was observed exclusively in denervated fibers, suggesting a pathophysiologic connection. Chronic denervation resulting from a dysfunctional neuromuscular junction has been reported in other cases of CMS12 [5], while a coincidental glycogen accumulation has not been identified so far. Although glycogen storage as consequence of denervation is not common, functional studies in experimental mouse models have demonstrated a metabolic shift and excessive glycogen accumulation in response to specific denervation processes [13, 14]. Hence, it might be speculated that the glycogen accumulation observed in this study represents a secondary phenomenon in the context of denervation.
Several studies have reported the presence of tubular aggregates as a main feature in CMS12 [2–5, 15–19]. In the histology of patient II.2, single myofibers contained small tubular aggregates, that were only identified in retrospect by thorough evaluation of serial sections. This is in line with other studies, reporting the subtle presence of tubular aggregates in some patients [3, 4]. Furthermore, abundant autophagic vacuoles were identified in ultrastructural analyses, confirming that impaired autophagy is a common feature of CMS12 [3, 6].
The clinical phenotype of the patients presented in this study is compatible with GFPT1-associated CMS in key aspects, although the clinical course of the siblings was rather severe. Although CMS12 typically manifests in the first decade of life, severe phenotypes with congenital onset, respiratory failure and bulbar involvement have been described in rare cases [2, 6]. Biallelic loss-of-functions variants have been identified in some severely affected individuals [3, 20]. However, congenital or infantile onset with delayed motor milestones has also been reported in children with biallelic missense variants [21]. Interestingly, one of the variants reported in these individuals (c.41G > T; (p.Arg14Leu)) involves the same amino acid affected in the present study (c.41G > A; (p.Arg14Gln)). Hence, it can be speculated that the severity of the clinical phenotype might not solely rely on the presence of truncating variants but also on the functional implications of the respective variant. Moreover, there was remarkable intrafamilial variety regarding the severity of symptoms and age of death in our family that cannot be explained. An altered susceptibility due to secondary predisposing factors by means of a genetic, epigenetic and posttranscriptional burden may be considered.
While patients with CMS12 usually show a similar pattern of muscular weakness, muscular atrophy, as seen in the siblings of our study, is rare [4]. On the other hand, myopathic changes detected in needle EMG studies are frequently observed in context of GFPT1-associated CMS [2]. These aspects, together with myohistology, were the reasons for primarily accounting the siblings’ phenotype to a genuine myopathy. Consequently, repetitive nerve stimulation was not performed at any time throughout the diagnostic procedure, neither in our department, nor in the departments that oversaw the siblings beforehand. Similar misclassifications have been reported for other patients with GFPT1-associatied CMS in previous studies [4]. Tragically, the siblings were unable to benefit from the diagnosis of a treatable hereditary disease, which was made post-mortem. However, the molecular diagnosis enabled a genetic counselling of the family. As the diagnosis of CMS12 was not made during the patients’ lifetime, disease-specific treatment with acetylcholinesterase inhibitors had never been initiated. This may have contributed to the severe course of the disease. Therefore, our case strongly emphasizes that evaluation of the decremental response should be included in the diagnostic standard for patients with unclear limb girdle syndrome to avoid missing a potentially treatable CMS.
In conclusion, the present study not only expands the spectrum of myohistological findings in GFPT1-associated CMS12 but also highlights the diagnostic value of NGS in regard to atypical or unsolved limb girdle syndromes. There is an immanent need for an integrative approach including thorough clinical workup, broad genetic analyses, and variant evaluation to overcome diagnostic pitfalls as evident in the present case.
Footnotes
ACKNOWLEDGMENTS
All authors thank the patients’ parents for their consent to participate in this study. Furthermore, we thank Dr. Astrid Pechmann for providing the clinical data of individual II.1 and Kathleen Zietz as well as Katrin Schulz for excellent technical assistance. T.B.H. was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) –418081722, 433158657.