
Abstract
This case report describes a girl who presented antenatal arthrogryposis and postnatal hypotonia, generalized and respiratory weakness, joint deformities particularly affecting the lower limbs and poor swallow. By 5 months, cataracts, abnormal electroretinograms, visual evoked potentials (VEPs) and global developmental impairments were recognized. No causative variants were identified on targeted gene panels. After her unexpected death at 11 months, gene-agnostic trio whole exome sequencing revealed a likely pathogenic de novo BICD2 missense variant, NM_001003800.1, c.593T>C, p.(Leu198Pro), confirming the diagnosis of spinal muscular atrophy lower extremity predominant type 2 (SMA-LED2). We propose that cataract, abnormal electroretinograms and VEPs are novel features of SMA-LED2.
INTRODUCTION
Arthrogryposis describes congenital contractures in two or more different joints and affects 1 in 3,000 live births. Over 400 conditions present with arthrogryposis; however, regardless of the specific cause, the underlying pathogenesis includes a reduction in fetal movements, leading to contractures [1]. Next generation sequencing now enables a genetic diagnosis to be reached in up to 60% of cases [2].
We describe an infant with antenatal arthrogryposis, post-natal hypotonia and cataracts, abnormal electroretinograms (ERGs), visual evoked potentials (VEPs) and global developmental impairments recognized over time. Gene-agnostic trio whole exome sequencing (WES) identified a de novo likely pathogenic variant in BICD2, which, to our knowledge, has not been described as causing cataract, ERG or VEP abnormalities.
CASE REPORT
This female baby was born at 36+2 weeks of gestational age to a healthy non-consanguineous Scottish couple. Antenatal ultrasound at 21 weeks revealed intrauterine growth restriction and arthrogryposis. She was delivered by elective caesarean section with Apgar scores of 5 and 8 at 1 minute and 5 minutes, respectively, and weighing 1.78 kg (–4.45 SDS). She had frontal bossing, upslanting palpebral fissures (Fig. 1A, B) and generalized hypotonia with reduced muscle bulk, particularly in the lower limbs. Finger creases were present and there was no dimpling over joints. Her shoulders were internally rotated with mild adduction deformities. She had mild elbow flexion contractures (10°) and restricted extension (to 150°). Her wrists could not be passively flexed beyond the neutral position. She had mild flexion deformities of proximal interphalangeal joints on the left and left fifth finger clinodactyly. Her hips were dislocated bilaterally, there was a flexion contracture (20°) of the right knee, bilateral vertical talus and ankle hyper-dorsiflexion bilaterally. There were little movements in facial muscles and flickers of movement only in the lower limbs, whilst upper limbs movements were part-range antigravity. She required extensive respiratory support, tracheostomy and percutaneous endoscopic gastrostomy due to unsafe swallow and gastro-oesophageal reflux.
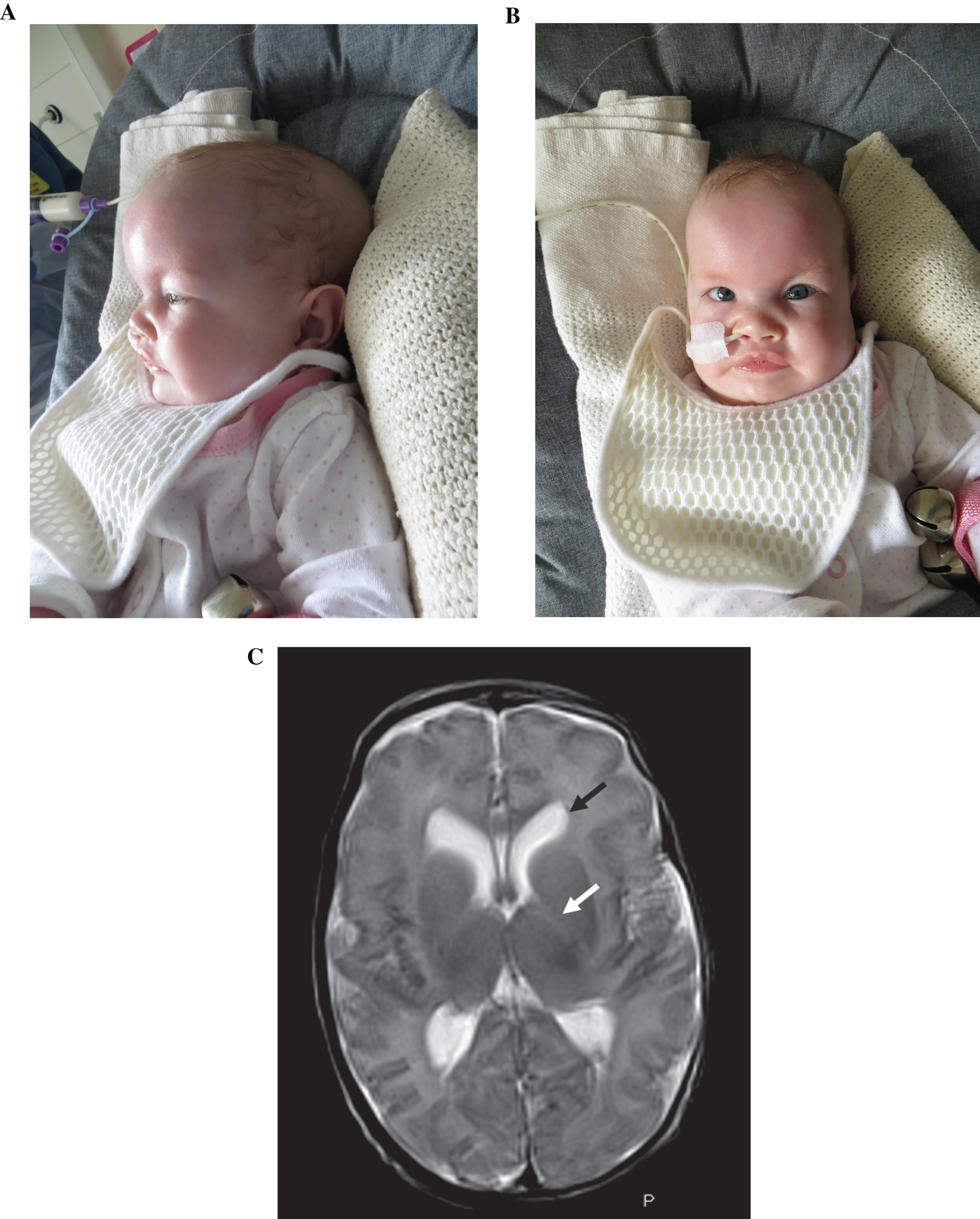
A, B. Clinical photographs aged 4 months. The patient has a frontal bossing (A) and upslanting palpebral fissures (B), the latter shared with her mother (not shown). Permission for publication of these photos has been granted by the parents of the patient. C. MRI Head: T2 sequence. There is absence of myelination in the posterior internal capsule (white arrow). There is splaying of the frontal horns of the lateral ventricles (black arrow).
Ophthalmology review at three months, delayed by illness, found exotropia, roving nystagmus and bilateral cataracts (dense lamellar on the left and more peripheral on the right). Ultrasound showed grossly normal features of both eyes and the dimensions were within normal range. VEPs at five months were present binocularly and monocularly, but with delay of the predominant peak; the right eye VEP was more complex and robust than the left eye VEP. ERGs under light-adapted conditions were present, but small and delayed with an atypical waveshape (Fig. 2). Given the electrodiagnostic results and the differences in the appearance of both cataracts, bilateral simultaneous surgery was performed. She was aphakic post-operatively with refractive correction initially provided with contact lenses and then prescription glasses. Visual impairment remained; however, her nystagmus lessened in frequency. There was light perception post-operatively in both eyes.
Over time, global developmental delay became evident. Although auditory brainstem evoked potentials were normal, she remained non-verbal with a pre-intentional level of communication. She did not achieve head control, nor independent sitting. After intensive physiotherapy, she was able to bring both hands together for finger play while supine. She developed flickers of hip adduction, abduction, and flexion.
A trial of pyridostigmine was performed without any clinical benefit. Nerve conduction studies were within normal limits (Supplementary Table 1).
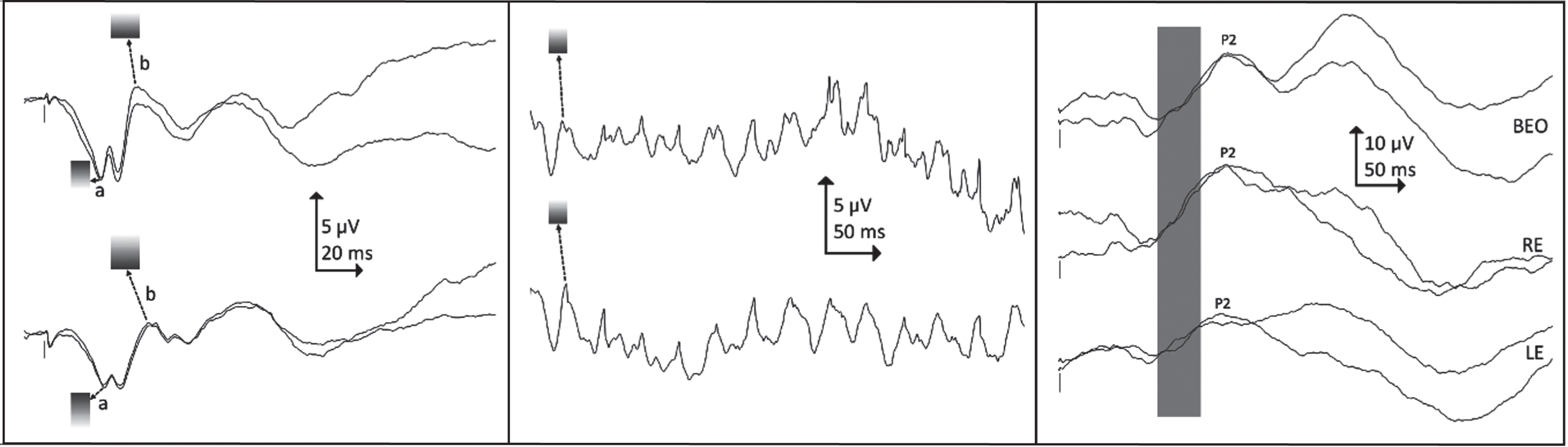
Visual electrodiagnostic findings. Left: light-adapted single flash ERGs, right eye above, left eye below. Shaded zones indicate reference limits for a- and b-waves: the patient’s ERGs are smaller and slower than normal, with a pronounced oscillation at the a-wave trough. Centre: light-adapted 30 Hz flicker ERGs, right eye above, left eye below: the patient’s ERGs are smaller than normal. Shaded zones indicate reference limits for flicker peak. ERGs were recorded during natural sleep using skin electrodes on the lower eyelid, with a high forehead reference (Fz). Reference limits have been adjusted to compensate for the known effect of closed lids. Right: binocular (top), right eye (middle) and left eye (bottom) flash VEPs. Shaded bar indicates normal timing for the predominant (P2) peak: the patient’s VEPs are delayed. VEPs were recorded during natural sleep, active electrode at Oz referred to Fz. The stimulus was a strong (40 cd s m-2) diffused xenon flash held a few centimetres from the closed eyelids. Positive is plotted upwards.
Creatinine kinase was normal (28 IU/L). There were no atypical transferrin glycoforms at 4.5 months. Brain MRI aged 1 month showed normal brain parenchyma, splaying of the frontal horns of lateral ventricles and absent myelination in the posterior limb of internal capsule consistent with delayed myelination due to prematurity (Fig. 1C). Echocardiography revealed a small apical ventricular septal defect.
She died unexpectedly at 11 months, due to refractory status epilepticus. Despite broad-spectrum antibiotics, antiviral therapy and inotropes she suffered two cardiac arrests. Her seizures failed to respond to appropriate, timely anti-seizure medication management and after discussion with her parents, active treatment was discontinued and she passed away peacefully.
Post-mortem findings were consistent with infection at the time of death. There was acute bronchopneumonia and positive norovirus antibody titres (Ct value 30). Cortisol levels were very high (>3,300nmol/l). Macroscopic examination of the brain was normal.
SNP-array showed a paternally-inherited 3.7 Mb deletion at 13q31.3-q32.1 containing eleven protein-coding HGNC genes, none associated with cataract. Only four of these were morbid genes, all for autosomal recessive disorders: CLDN10 (Helix Syndrome, OMIM #617671), DNAJC3 (cerebellar and peripheral ataxia with hearing loss and diabetes mellitus, OMIM #616192), GPC6 (omodysplasia type 1, OMIM #258315) and TGDS (Catel-Manzke syndrome, OMIM #616145). She tested negative for the myotonic dystrophy type 1 repeat expansion, and sequencing of congenital myasthenic syndrome genes revealed no causative variants. Testing of a clinical exome panel of 107 neuromuscular arthrogryposis genes was initiated and customized, when the cataract was recognized, to include genes known to cause both cataracts and arthogryposis (ALG14, ALG3, COG6, COG7, COG8, DPACGT1, DYNC1H1, ERCC5, HRAS, INPP5K), but no causative gene variants were found. She was heterozygous for a pathogenic LAMA2 variant c.2049_2050del, p.(Arg683Serfs*21)). After her death, trio WES using an inheritance-based, gene-agnostic approach revealed a heterozygous de novo BICD2 missense variant (NM_001003800.1, c.593T>C, p.(Leu198Pro)), confirmed on Sanger sequencing and classified likely pathogenic (ACMG criteria: PS2 moderate, PM2 moderate, PP3 supporting, PP4 supporting [3]). Analysis of the PanelApp Cataracts version 2.3 gene panel was applied and excluded a reduced penetrance heterozygous inherited variant [4].
DISCUSSION
Dominant variants in BICD2 (BICD Cargo Adaptor 2, #OMIM 609797) cause spinal muscular atrophy, lower extremity-predominant type 2 (SMA-LED2, #OMIM 615290) [5–7]. Clinical severity varies and includes severe, antenatally lethal forms, congenital forms with or without contractures, and childhood and adult-onset lower extremity weakness [8–11]. De novo variants in BICD2 have been specifically shown to be correlated with more severe clinical features including arthrogryposis and ealy fatal outcome [8, 12].
Retrospectively, the disparity between our patient’s severe lower limb weakness and much milder upper limb weakness is characteristic of SMA-LED and similar disparity is evident in her neurophysiology, where CMAPs were much smaller in lower than upper limbs, despite being within normal limits for age (Supplementary Table 1).
Upper motor neuron involvement and neurocognitive involvement were highlighted in the initial description of SMA-LED2 [6]. Subsequently, cerebellar hypoplasia and perisylvian polymicrogyria were described [10]. Although no structural brain changes were identified in our patient, we presume her neurodevelopmental impairment relates to central involvement.
Our patient’s abnormal light-adapted single flash ERGs were similar in morphology to those seen in congenital disorder of glycosylation type Ia (CDG1a, OMIM #212065) [13], however is unlikely that this child had co-existing CDG1a because her neurological features were atypical, transferrin glycoforms were normal and no pathogenic variants in PMM2 (OMIM #601785) were identified on WES.
We propose that cataract and abnormal retinal function may be features of BICD2-related disorders, as no alternative cause of these was found on extensive genetic testing. Furthermore, early onset cataracts have been described in patients with variants in DYNC1H1 (OMIM #600112) [14], which encodes the cytoplasmic heavy chain 1 of dynein, an important binding partner of BICD2. DYNC1H1 variants cause a range of phenotypes including SMA-LED1 [14, 15], which is clinically strikingly similar to SMA-LED2 [11], therefore it seems reasonable that this similarity could extend to include cataract, especially since our patient’s variant lies in the BICD2’s dynein binding domain (CC1) [16–18]
Moreover, as a golgin and cargo adaptor protein [7], linking the dynein-dynactin motor complex with its intracellular cargo [18], BICD2 plays a role in retrograde axonal transport, vesicular transport, Golgi integrity and synaptic vesicle recycling [7], any or all which could be affected by our patient’s variant.
Although no studies have directly shown that, both DYNC1H1 and BICD2 are expressed in the retina [20] and during retinal development [21]. DYNC1H1 is expressed in the elongating fiber cells of rats’ lens where it is suggested to play a role in trafficking of vesicles along microtubules during development [19] and a non-sense DYNC1H1 variant has been shown to impair photoreceptor development in zebrafish [22]. The role of Bic-D in microtubule-based transport was also shown to be essential for photoreceptor nuclear migration during eye development in Drosophila [23]. A role for BICD2 in eye development is further suggested by the finding that another of its binding partners, Rab6 [24], is involved in trafficking rhodopsin in post-Golgi vesicles during photoreceptor development and maintenance [25, 26].
Finally, since eye involvement has not been described in SMA-LED2 before, it may be a rare feature. It is possible that in more severely affected cases their clinical severity has precluded detailed formal assessment.
The intractable seizures at the end of our patient’s life were unexpected and occurred in the context of a pyrexial illness. Although seizures have been described in three other SMA-LED2 cases, two had structural brain malformations and one had severe ischaemic brain injury [8, 12]. Seizures have also been described in patients with DYNC1H1 variants [14, 28]. Our patient’s seizures could be entirely unrelated to the SMA-LED2 or reflect a non-specifically reduced seizure threshold because of her central involvement. Nonetheless, it is notable that the seizures were resistant to standard therapy and it may be reasonable to maintain a low clinical index of suspicion for seizures in SMA-LED2.
The rapid terminal decompensation in our patient could raise the possibility of a second, metabolic, disorder [29]. However, neither biochemical testing, nor WES found any evidence for this.
This patient had extensive genetic testing in life, with only a heterozygous pathogenic LAMA2 variant detected, which is presumed to represent incidental carrier status since her clinical phenotype and normal CK make merosin-deficient congenital muscular dystrophy (MDC1A, OMIM #607855) most unlikely [30]. This illustrates the difficulty in selecting targeted gene panels in cases where the clinical phenotype is still evolving and highlights the utility of gene-agnostic trio WES as a first line investigation in such cases.
In conclusion, we believe this is the first report of eye involvement in SMA-LED2 and suggest that cataract, abnormal ERG waveforms and abnormal VEPs may be features of the condition in some cases.
Footnotes
ACKNOWLEDGMENTS
We would like to sincerely thank the parents for very kindly allowing us to publish the details of their daughter. We would also like to thank the Glasgow Regional Genetic Laboratory who performed the SNP array and the Exeter Genomic Laboratory for the whole exome analysis.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to report and this research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.