
Abstract
Background:
Anti-3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR)-myopathy is a usually rapidly progressive form of immune-mediated necrotizing myopathy (IMNM). Rarer clinical courses show slow progression and resemble the phenotype of limb-girdle dystrophy (LGMD).
Objective:
We demonstrate the difficulties in differentiating LGMD versus anti-HMGCR-myopathy.
Methods:
We report on a 48-year-old patient with slowly progressive tetraparesis and hyperCKemia for more than 20 years.
Results:
Due to myopathic changes in initial and second muscle biopsy and typical clinical presentation, the patient was diagnosed with LGMD 20 years ago; despite comprehensive genetic testing including exome diagnostics, the genetic cause of disease could not be identified. Finally, HMG-CoA reductase antibodies were detected, confirming the diagnosis of anti-HMGCR-myopathy. By re-work-up of a second muscle biopsy specimen from year 2009, the diagnosis of a IMNM was made in retrospect. Seven cycles of high-dose immunoglobulins were administered; patient reported outcome measures have mildly improved.
Conclusion:
Patients with clinical LGMD phenotype, degenerative changes in muscle biopsy but without genetic confirmation of the disease should be tested for HMG-CoA-myopathy, thereby allowing for an early start of treatment.
INTRODUCTION
Inflammatory myopathies are a heterogeneous group of diseases consisting of dermatomyositis, polymyositis, immune-mediated necrotising myopathy, and inclusion body myositis. Only in 2004, immune-mediated necrotising myopathy (IMNM) was identified as a new entity, defined by autoantibodies against signal-recognition peptide (SRP) and 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) [1, 2]. IMNM usually presents with acute to subacute proximal tetraparesis, myalgia and hyperCKemia; in some patients, dysphagia may be observed in addition [2–4]. Patients with anti-SRP IMNM usually show a clinically more severe course than patients with anti-HMGCR-antibodies [1]. HMGCR-IMNM is frequently (35%) associated with the previous use of statins [5–7]. However, many patients have no identifiable risk factors [5]. Interestingly, mushrooms are a naturally occurring source of statins, which might be able to initiate anti-HMGCR antibodies at low titers [8].
The detection of autoantibodies is pathognomonic in diagnosis of IMNM. However, it must be noted that there are numerous cases of seronegative IMNM, which show significantly higher rates of additional extramuscular disease activity (interstitial lung disease, arthritis, Raynaud phenomenon, skin or cardiac symptoms) [9]. Considering seropositive IMNM, electromyography shows myopathic changes, often associated with pathological spontaneous activity. Routine muscle biopsy demonstrates scattered necrotic muscle fibres in different stages with limited inflammatory infiltrates. Necrotic fibres are anti-C5B9 complement positive and might display anti-p62 staining [10]. In addition to glucocorticoids, a wide variety of immunosuppressive and immunomodulatory treatments are used [3].
Apart from the acute form, there are patients with a milder and slowly progressive course, phenotypically mirroring limb girdle muscular dystrophies (LGMD) [2, 11–13]. LGMDs are a group of muscular dystrophies with a wide variety of clinical and genetic subtypes, characterised by a slowly progressive, predominantly proximal limb-girdle weakness. Calf hypertrophy is frequently observed, while facial weakness is rare [14].
We demonstrate the difficulties in differentiating LGMD versus anti-HMGCR-myopathy in the following case report.
CASE REPORT
We report on a 48-year-old patient with slowly progressive limb-girdle weakness who was misdiagnosed with LGMD for two decades; 20 years later, in 2020, IMNM with anti-HMGCR-antibodies was diagnosed (Fig. 1).
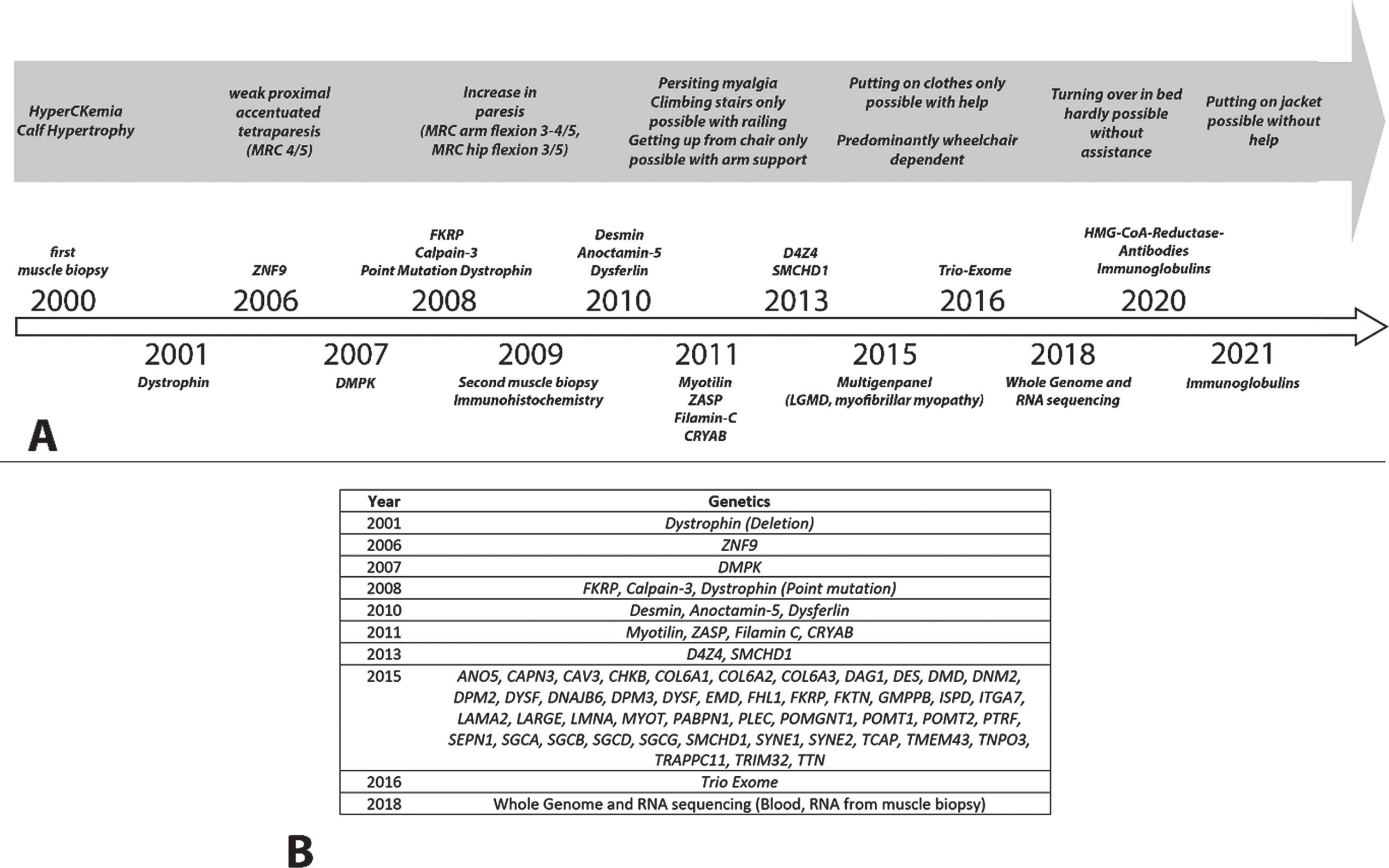
Flow-chart of the clinical disease course (A) and the conducted genetic testing (B).
In the year 2000, an elevated creatine kinase (CK) (2300 U/l, normal upper limit 180 U/L), was found during routine work-up. At that time, a mild calf hypertrophy without additional muscle weakness was detected. Muscle MRI displayed signal enhancement in the iliopsoas muscles, while electromyography showed a normal pattern. A muscle biopsy of the right gastrocnemius muscle showed an increase in fibre size variability with smaller and round fibres. Furthermore, the biopsy revealed a limited increase of perimysial connective tissue and a few necrotic fibres, comparable with the diagnosis of a degenerative myopathy. Immunoblotting for dystrophin showed a band with reduced molecular weight and normal amount of dystrophin; however, dystrophinopathy due to deletions, duplications and point mutations in the dystrophin gene was genetically excluded later on. Due to idiopathic hearing loss, the patient received intravenous steroids. A significant decrease in CK levels to 800 U/l was seen under steroid therapy.
In 2006, neurological examination showed a progressive weakness in thigh (MRC 4/5) and shoulder girdle muscles (MRC 4-5/5) with marked muscle atrophy. Climbing a staircase was impaired, carrying heavy items was no longer possible. Since he and his wife expected their first child, further genetic testing was initiated; family history did not reveal any neuromuscular diseases. Genetic testing for myotonic dystrophy type 1 and 2 was negative, although complex repetitive discharges and myotonic runs had been evident in EMG. A follow-up muscle MRI showed a fatty atrophy of the thigh muscles without edema or contrast enhancement. Laboratory testing for ANA, ENA, anti-Jo1, anti-SRP, and anti-Mi-2 antibodies was normal. Meanwhile, CK levels had been elevated up to 8000 U/l and the patient additionally reported on generalized myalgia. With progressive disease, bilateral weakness of arms (MRC 3-4/5) and hip flexors (MRC 3/5) was evident. There was no cardiac or respiratory involvement, no scoliosis and no scapular winging. Alpha-glucosidase enzyme activity testing in leukocytes showed no evidence of Pompe disease.
In 2009, a second muscle biopsy of the left vastus lateralis muscle showed an increase in fibre size variability with smaller and round fibres. An increase of perimysial connective tissue was not found but a limited inflammation preferably by anti-CD68 and anti-CD4 positive stained cells, and scattered necrotic fibres in different stages. Immunohistology additionally showed a sarcolemmal MHC1 upregulation and scattered positive anti-C5B9 and anti-p62 stained necrotic fibres. Immunohistochemical stainings also showed a markedly reduced staining for dysferlin, reduced staining for dystrophin (Fig. 2), while adhalin, merosin, caveolin-3, emerin and alpha-dystroglycan were normal. The decrease of dysferlin protein is most likely related to the inflammation, as this secondary reduction is a common finding in e.g. inflammatory myopathies. Normal immunoblotting excluded dystrophinopathy (this time with a normal band and amount of the dystrophin protein), calpainopathy (LGMDR1) and dysferlinopathy (LGMDR2). Further genetic testing was unremarkable (Fig. 1B).
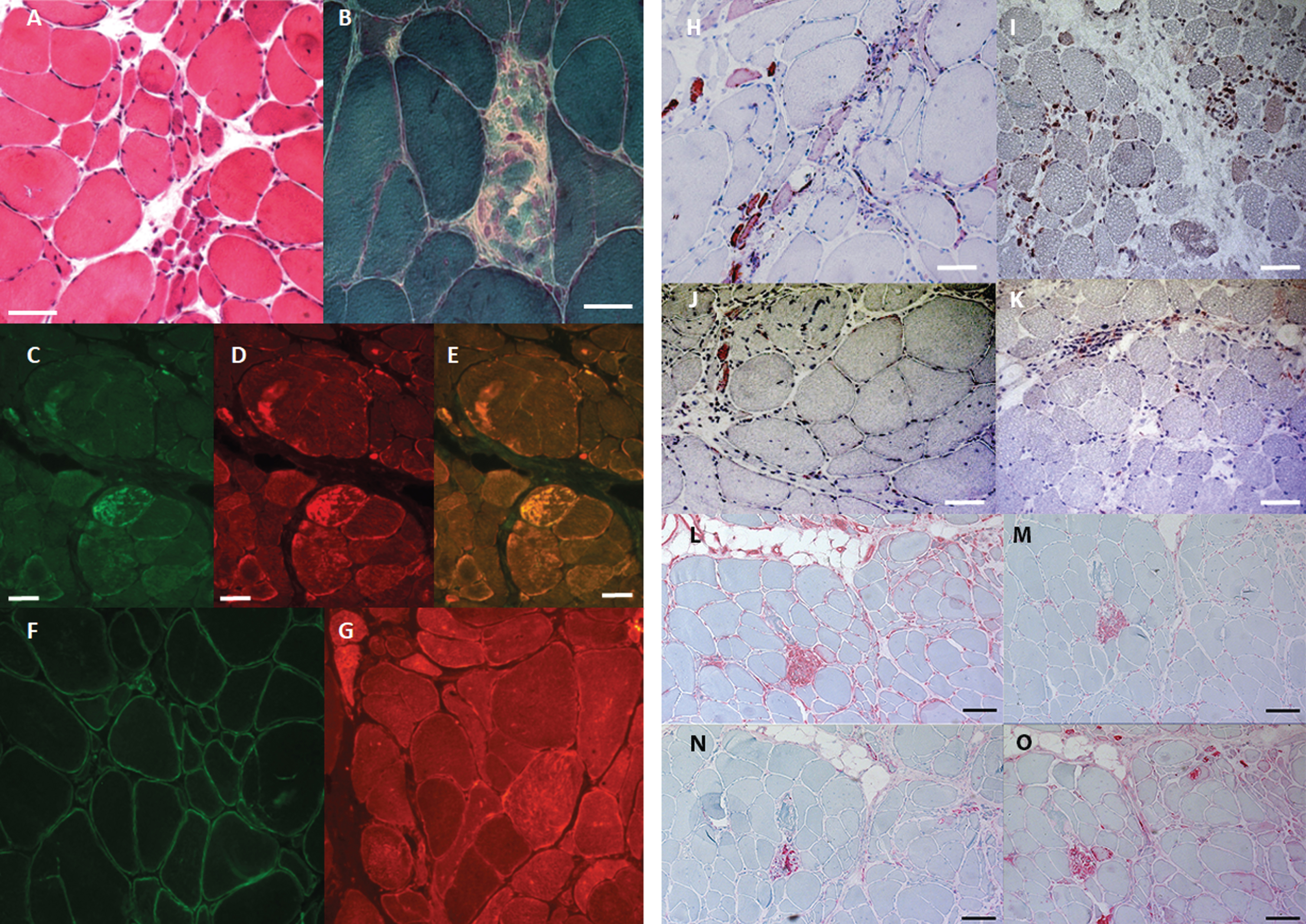
Histology: Biopsy specimen taken from the left vastus lateralis muscle at the age of 36 years, showed (A, H&E) a pronounced fibre size variability, fibre splitting, small groups of atrophic fibres, and centralized nuclei in hypertrophic fibres. (B): Trichrome Gomori showed scattered myofiber necrosis. (C-E: serial sections) Immunohistology revealed intracellular aggregation of myofibrillar proteins (C) anti-desmin, (D) anti-aB-crystallin (E) overlay. (F-G: serial sections) Anti-dystrophin (F) and anti-dysferlin stainings showed marked reduction of both muscle membrane proteins. Size bars in A-F adjusted to 50μm. (H-I) Anti-BCL-2 immunostaining labels positive scattered atrophic fibres and lymphocytic infiltrates. (J-K) Anti CCR-4 immunostaining shows only limited lymphocytic staining. (Bars in H-K adjusted to 50μm). (L) Anti-MHC1 staining presented a sarcolemmal upregulation. (M) Anti-CD4 staining showed several positive lymphocytes. Anti-C5B9 staining (N) and anti-p62 (O) revealed scattered complement activation and positive diffuse anti-p62 in necrotic fibres in different stages.
In 2010, there was significant worsening of muscle weakness. Climbing stairs was only possible with railing, getting up from a chair with the help of the arms. Constant muscle soreness-like, dragging pain was still present. Steroid treatment with Deflazacort was initiated with 90 mg/d for 15 months and slowly reduced to 42 mg/d over 6 months. During steroid treatment, hip flexion improved to MRC 4-5/5. Treatment was continued with 42 mg/d over 4 years with considerable side effects (cataract, onset of Cushing’s disease). However, paresis of foot extensors, foot flexors and axial weakness developed in 2013.
Subsequently, we genetically tested for anoctaminopathy (LGMDR12) without pathogenic findings. Dystrophinopathy due to deletions, duplications and point mutations in the dystrophin gene was excluded by MLPA and NGS analysis, along with negative testing for facioscapulohumeral muscular dystrophy type 1 and 2.
In 2015, free walking was only possible assisted for short distances, and the patient became wheelchair dependent and needed help for dressing himself, still under treatment with 42 mg deflazacort daily. A whole-body muscle-MRI for pattern recognition showed a symmetrically distributed degeneration of the skeletal muscles, especially in the upper extremities, trunk and thighs. The pelvic muscles were particularly affected as well as the hamstring muscles (Fig. 3).
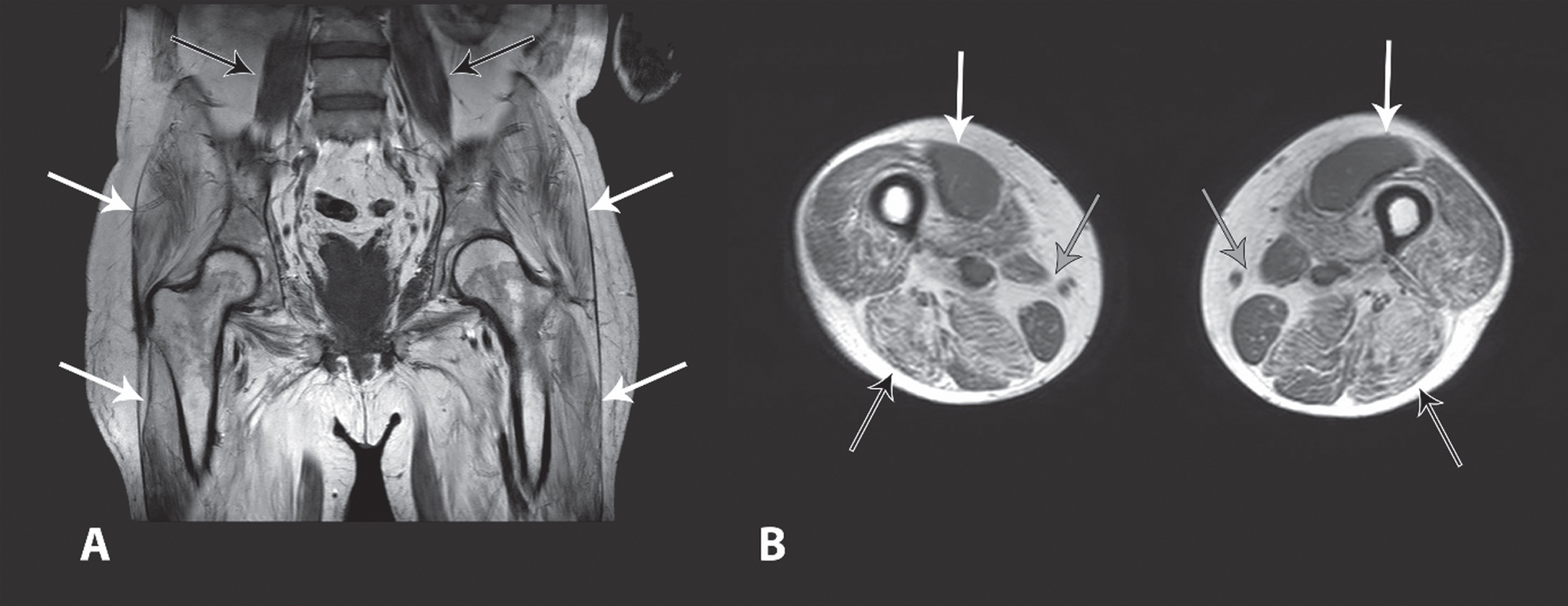
Whole-body muscle MRI of the patient: (A) Pelvis: In the T1-weighted nonfat-saturated T1-weighted sequence in coronal section, prominent fatty degeneration of the entire pelvic musculature was detected (white arrows); only the pelvic floor muscles on the left are not as clearly affected. Regular view of the psoas muscle is displayed on both sides (black arrows). (B) Transverse thigh: T1-weighted sequence without fat saturation in axial section showed marked fatty degeneration of the dorsal thigh muscles (black arrows), especially the M. biceps femoris, M. semimembranosus, and less prominent M. semitendinosus. Additionally, compared to the hamstring musculature less extensive fatty degeneration of the M. quadriceps femoris, in particular the M. vastus medialis (white arrows) were well preserved in the ventral part. The M. gracilis, the M. sartorius and the M. adductor magnus also appeared less reduced in volume or by fatty degeneration (grey arrows). Permission to publish this figure has been granted by the patient.
Still, no LGMD subtype could be assigned. Multigene panel diagnostics for LGMD and myofibrillar myopathies (Fig. 1B) showed a single heterozygous variant of unknown significance in each TRIM32 and SYNE2. Follow-up trio exome sequencing of the patient and his parents revealed the SYNE2 variant in the healthy father of the patient, excluding EDMD5. No further variants of interest were found.
In 2018, getting up from a chair or turning in bed was no longer possible without help. Following transfer from car to wheelchair, the patient suffered a fall along with a lumbar spine fracture and concussion. Therefore, Deflazacort was reduced to 6 mg/d followed by worsening of hip flexor strength to 2/5 bilaterally.
In 2016, re-analysis for trio exome data was initiated, and from 2018 to 2020 WGS from EDTA blood and RNAseq from RNA isolated from the muscle biopsy was performed, both without any striking candidate genes.
Following the paper of Mohassel et al. [11], elevated anti-HMGCR-antibodies were identified with > 200 U/ml (normal < 20 U/ml). A significant elevation of anti-HMGCR antibody-titer was confirmed by ELISA independently (Mammen laboratory), while IP analysis was equivocal. However, the patient had never been treated with statins.
Kurashige et al. reported a higher incidence of bcl2- and CCR4-positive lymphocytes in muscles of anti-HMGCR-IMNM patients [15]. Therefore, these stainings were performed on the muscle specimen from the year 2009, showing bcl2-positive endomysial and perivascular infiltrates. Interestingly, regenerating muscle fibres were also positive for bcl2 (Fig. 2H-I) as previously reported [16]. Furthermore, CCR4-positive perimysial and endomysial lymphocytes were observed (Fig. 2J), partially in lymphocytic accumulations (Fig. 2K) as has been discussed earlier. Moreover, the scattered necrotic fibres were anti-C5B9 complement positive and revealed a diffuse anti-p62 staining, comparable to findings by Fischer et al. for HMGCR associated IMNM [10].
Anti-HMGCR-myopathy was finally diagnosed, and the patient was treated with seven cycles of high-dose intravenous immunoglobulins (0.4 mg/kg body weight). Deflazacort was increased to 60 mg/d and afterwards tapered to 6 mg over 4 months. The initial steroid treatment led already to a reduction of the CK (1291 to 443 U/l) and the concentration of the anti-HMGCR-antibody (> 200 to 88 U/l), confirming the steroid responsiveness noted in the patient’s past history. Under IVIG treatment the CK stabilised around 280 U/l. Still, marked weakness of 3/5 in the proximal upper extremities, and 2/5 in the proximal lower extremities remained. However, the patient reported outcome measures such as enhanced stability in standing have improved, and the patient is again able to put on a jacket independently.
DISCUSSION
Keeping in mind that HMGCR-antibodies were first described in 2004, we report on a 48-year-old patient with anti-HMGCR-myopathy who was misdiagnosed with LGMD for 20 years. During the course of the disease, he slowly developed severe limb-girdle weakness, with leg weakness starting earlier and progressing more severely compared to arm muscle weakness, and started using a wheelchair from age 42.
The slow progression and the initially observed mild calf hypertrophy are not a typical finding in anti-HMGCR-myopathy; however, there are several other patients presenting with an LGMD phenotype [2, 18]. Mohassel et al. detected anti-HMGCR-antibodies in six out of 128 patients with a former diagnosis of hereditary myopathy. Patients with a CK > 1000 U/l, limb-girdle weakness, histologic findings of degenerative myopathy, no other affected family members and a distinct imaging pattern (posterior thigh/hamstring muscles more affected than the anterior thigh/ventral proximal muscles) were included [11]. These criteria might be helpful in differentiating patients with anti-HMGCR-myopathy from LGMD patients. However, there are many LGMDs / hereditary myopathies with a predilection for the posterior compartment of thigh (e.g. CAPN3, FKRP, Laminin alpha 2, Emerin, PNPLA2, DNAJB6, FSHD, OPMD). Therefore the criteria are rather helpful to highlight those patients within an LGMD cohort that might have HMGCR, in particular if negative by genetic testing. However, it should be noted that anti-SRP antibodies can present with a LGMD-like phenotype, especially in young patients, and should therefore be considered as well [20].
In addition, a diagnosis of IMNM cannot be made solely by testing for antibodies, considering the group of seronegative IMNM. With regard to the distinction of LGMD and IMNM, seronegative IMNM can possibly be excluded due to the high frequency of extramuscular symptoms (up to 50% of patients according to Lim et al.) [9].
Our patient showed similar findings as described by Mohassel et al. –no affected family members, high CK, myopathic muscle biopsy –however, clinically, predominant posterior compartment leg muscle weakness was not seen initially, although hamstring muscle involvement was identified in the MRI scan. Later in the disease course, posterior weakness became more pronounced. In two muscle biopsies, myopathic findings were seen along with scattered necrotic fibres in different stages, consistent with the findings of muscle biopsies of similar cases [5, 18]. Unfortunately, we did not have the possibility to reevaluate the muscle biopsy from the year 2000. However, fibre necrosis was not a predominant finding in our patient in the description of the first biopsy, but was more pronounced in the second muscle biopsy from the year 2009. In the recent work-up of the patient, the immunohistology of the 2009 biopsy showed anti-p62 and anti-C5B9 complement positive necrotic fibres, supporting the diagnosis of IMNM. Immunohistochemistry repeatedly showed reduced staining for dystrophin and dysferlin –while dysferlin reduction, in particular with increased intracellular staining, has been widely shown to be a rather nonspecific finding in necrotizing myopathies, while truncated dystrophin reduction has not yet been described, but was seen only in the first biopsy, so this finding remains unclear as most authors reported on normal immunohistochemical findings [5, 12]. Extensive molecular testing including trio exome and WES was performed over the years, without revealing striking candidate genes. However, LGMD can be genetically confirmed in only 40-60% of cases [21]; therefore, lack of a definite genetic diagnosis does not necessarily exclude LGMD. Anti-HMGCR-myopathy might also co-exist with a hereditary myopathy, although no such cases have been reported to our knowledge [22]. Nevertheless we cannot completely rule out a co-existence in our patient.
MRI showed symmetric atrophy and fatty degeneration of pelvic and thigh musculature in our patient, predominantly in the hamstrings muscles, similar to Mohassel et al. [11] and Idiculla et al., the latter with more asymmetric distribution [17]. The paraspinal and gluteal muscles were also affected in our patient, as described by Mohassel et al [11].
Bcl2-specific lymphocytes, upregulation of Bcl2 in regenerating fibres as well as CCR4-positive lymphocytic infiltrates and accumulations and the anti-C5B9 and anti-p62 staining pattern suggested an anti-HMGCR-myopathy. Anti-Bcl2, anti-CCR4, and anti-p62 may represent relevant biomarkers to distinguish hereditary myopathies from immune-mediated necrotising myopathies and can further support the diagnosis of anti-HMGCR-myopathy.
In our patient, the positive response to steroid treatment was the main reason to revisit the diagnosis. Steroids have been reported in several case studies or small patient cohorts to be beneficial in some [23–25] but not all [26] LGMD subtypes, and a treatment attempt can be justified on an individual case-by-case basis. Our patient had been treated with steroids from an early stage of disease, first in an intermittent, later on in a permanent treatment regimen, in an attempt to slow down worsening of muscle strength and due to an initial positive response to steroid treatment. With 42 mg Deflazacort daily, our patient showed a 10-fold decrease in CK –which is frequently seen during steroid treatment and not necessarily associated with improvement of the underlying disease [11] –and improvement of hip flexion strength from 3/5 to 4-5/5. However, dose reduction to 6 mg/d following the LWK1 fracture resulted in worsening of hip flexion to 2/5.
Mohassel et al. [11] reported on clinical improvement in all patients, more pronounced in paediatric patients with shorter disease duration, but also in two patients with a disease duration of 20 years. One wheelchair-dependent patient even started walking again with help of a rolling walker.
We could not see marked improvement of strength after seven IVIG cycles, but stabilisation and mild improvement of patient reported outcomes such as enhanced stability in standing and dressing independently. The limited response is most likely related to the fatty degeneration and atrophy of proximal muscles resulting in severe functional disability prior to treatment initiation.
Mohassel et al. [11] treated the former wheelchair-dependent patient with high-dose steroids along with IVIG (IVIG 1.5 g/kg plus 750 mg methylprednisolone every 3 weeks), which could not be reproduced in our patient due to steroid side effects (see above). Others reported on non-responders to additional immunosuppressive treatment, and suggested an early start of Rituximab in the treatment regimen, not only in LGMD-like cases [17, 27]. Due to the longstanding disease with fatty replacement in the MRI and lack of edema, we decided against Rituximab treatment; however, this may be an additional future treatment option.
Overall, early start of immunosuppressive treatment is essential since progressive loss of muscle along with fatty replacement cannot be reversed and may lead to poor outcome. Nevertheless, escalation of immunosuppressive and immunomodulatory treatment is warranted. Various studies show the best therapeutic benefit using methotrexate and IVIG in a prednisone-refractory cohort [7]. Other agents such as mycophenolate, cyclophosphamide, cyclosporine and azathioprine are also used with or without corticosteroids [28–31] and could –apart from Rituximab - possibly represent future treatment options for our patient.
Our patient highlights the diagnostic challenges in differentiating LGMD and anti-HMGCR-myopathy and reveals possible diagnostic pitfalls, when a non-confirmed diagnosis is not revisited. Importantly, early start of therapy is crucial for a favourable clinical outcome.
Additional to muscle biopsy and electrophysiology, testing for anti-HMGCR-antibodies (as well as anti-SRP-antibodies) should be included in the diagnostic work-up of LGMD, even if the clinical diagnosis of LGMD seems unequivocal. Vice versa, muscle pathology resembling IMNM may be encountered in LGMD awaiting genetic diagnosis, so both entities should always be considered.
FINANCIAL DISCLOSURES
Maggie C. Walter has served on advisory boards for Avexis, Biogen, Novartis, Pfizer, Roche, Santhera, Sarepta, Pharnext, PTC Therapeutics, Ultragenyx, Wave Sciences, received funding for Travel or Speaker Honoraria from Avexis, Biogen, PTC Therapeutics, Ultragenyx, Santhera, Sarepta, and worked as an ad-hoc consultant for AskBio, Audentes Therapeutics, Avexis, Biogen Pharma GmbH, Fulcrum Therapeutics, GLG Consult, Guidepoint Global, Gruenenthal Pharma, Novartis, Pharnext, PTC Therapeutics, Roche.
Sabine Krause has served on an advisory board for Ultragenyx and has an MTA with Daiichi Sankyo.
Benedikt Schoser has served on advisory boards for Argenx, Amicus, Astellas, Lupin, Sanofi Genzyme, Spark, Tysha; Contracted research: Amicus, Merck; Honoraria for lectures: Kedrion
Miriam Hiebeler, Carsten Bönnemann, Payam Mohassel, Andrew Mammen, Katherine Pak, Maria Ingenerf and Raimo Franke have no Conflicts of Interest.
Footnotes
ACKNOWLEDGMENTS
We thank Ursula Becker for expert technical assistance. We thank H. Saodiy, I. Hirschmann and F. Walter for scientific assistance and database research.