
Abstract
Background:
Proximal muscle weakness may be the presenting clinical feature of different types of myopathies, including limb girdle muscular dystrophy and primary mitochondrial myopathy. LGMD1B is caused by LMNA mutation. It is characterized by progressive weakness and wasting leading to proximal weakness, cardiomyopathy, and hearth conduction block.
Objective:
In this article, we describe the case of a patient who presented with limb-girdle weakness and a double trouble scenario –mitochondrial DNA single deletion and a new LMNA mutation.
Methods:
Pathophysiological aspects were investigated with muscle biopsy, Western Blot analysis, NGS nuclear and mtDNA analysis and neuromuscular imaging (muscle and cardiac MRI).
Results:
Although secondary mitochondrial involvement is possible, a “double trouble” syndrome can not be excluded.
Conclusion:
Implication deriving from hypothetical coexistence of two different pathological conditions or the possible secondary mitochondrial involvement are discussed.
INTRODUCTION
LMNA-related disorders, or laminopathies, are rare diseases mostly caused by mutations in the LMNA gene, which encodes for the nuclear envelope proteins, lamin A and C, via alternative splicing. Different phenotypes with skeletal muscle involvement have been linked to LMNA mutations, with often shared clinical aspects thus defining a broad phenotypic continuum [1, 2]. Among these myopathic forms, LGMD1B (also define LMNA-EDMD, accordingly to the new proposed LGMDs nomenclature [3]) is characterized by proximal muscle weakness associated with left ventricular enlargement, conduction system disease and arrhythmias [1]. Its clinical and genetic diagnosis is crucial for cardiac management and genetic counselling. Mitochondrial DNA single deletion is a sporadic disorder associated with a spectrum of different clinical phenotypes, ranging from chronic progressive external ophthalmoplegia (PEO), to PEO-plus phenotype (PEO associated with proximal myopathy, dysphagia, respiratory failure) and the full-blown Kearns-Sayre syndrome [4]. Here, we describe a patient presenting with a double trouble scenario, carrier of both a new LMNA gene variant (nucleotide insertion with frameshift) and a mitochondrial DNA single deletion.
MATERIALS AND METHODS (CASE PRESENTATION)
A 63-year-old man arrived to our attention for a one-year history of muscle cramps, exercise intolerance, progressive proximal weakness at lower limbs and mild hyperCKemia (400 UI/L). He had a history of hypertension lasting 10 years. Blood tests showed a mild impairment in renal function with glomerular filtration rate (eGFR) of 47 mL/min. The patient reported that the father was suffering from a progressive weakness of the lower limbs, starting at the same age, and died at 83years old from stroke.
The neurological examination showed asymmetric winged scapula and femoral quadriceps wasting with bilateral motor deficit in leg extension and hip flexion (muscle strength valued with Medical Research Council Scale (MRC) of 4/5) (Fig. 1). The present study conforms to the ethical standards and guidelines of the journal.
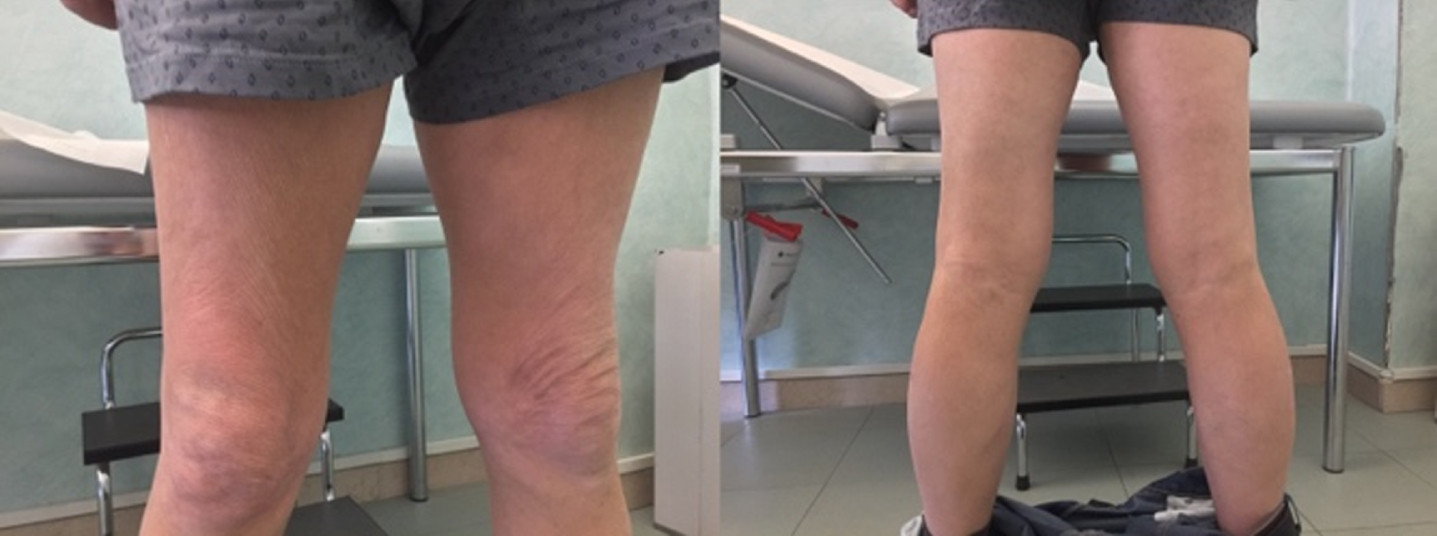
Clinical examination, showing wasting of thigh muscles, especially distal quadriceps.
The patient has given written and informed consent for online publication of his clinical information.
RESULTS
The electromyography revealed a myopathic pattern with spontaneous activity (fibrillation potentials) in tibialis anterior muscles. Myositis specific antibodies were negative. A forearm ischemic test detected basal hyperlactacidemia (basal lactate 29,8 mg/dl) and increased lactate production after exercise. Muscle MRI showed involvement of gastrocnemius medialis and thigh, with quadriceps fatty infiltration and oedema (Fig. 2). The deltoid muscle biopsy revealed mitochondrial abnormalities, in particular scattered ragged-red fibres and several COX negative fibres (Fig. 3), fibre degeneration with myofagocitosis and focal inflammatory infiltration. Mitochondrial DNA long-range PCR analysis disclosed a single deletion in the mtDNA, from 7061 bp to 13613 bp, with a 15%heteroplasmy rate (Fig. 4). Moreover, the NGS analysis for limb girdle muscle dystrophies/myopathies genes panel identified a previously unreported heterozygous variant in exon 11 of the LMNA gene (c.1861dupA, p.Thr621Asnfs*83). The variant is predicted to disrupt protein reading frae and to determine the loss of the original stop codon with formation of a longer protein. In silico predictions supported its pathogenicity.
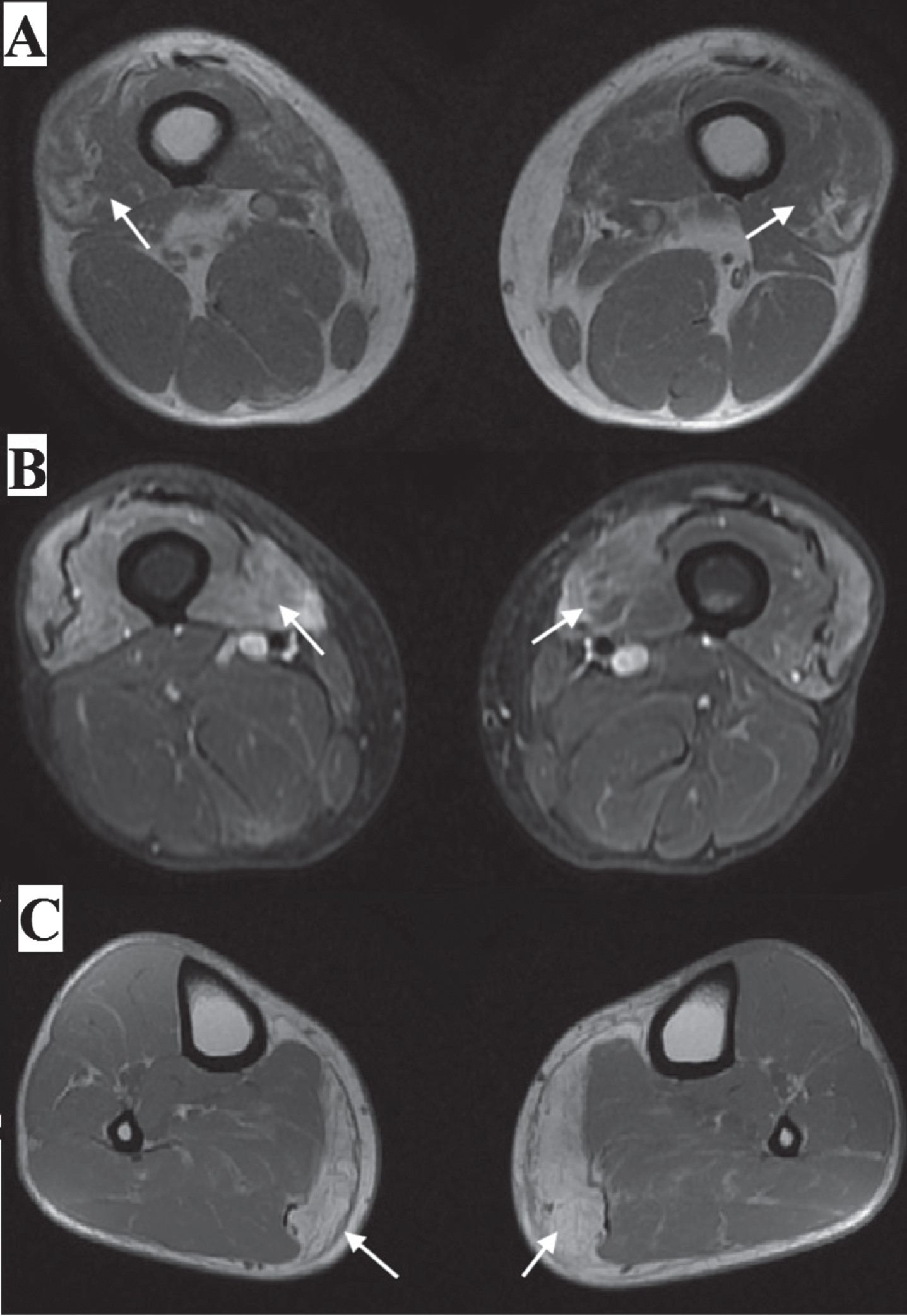
Muscular MRI, showing distal quadriceps atrophy and edema (arrow): (A) axial in-phase/out-of-phase gradient echo sequences (GRE); (B) short-tau inversion-recovery (STIR) sequence: (C) leg MRI, showing the prominent involvement of gastrocnemius medialis (arrow).
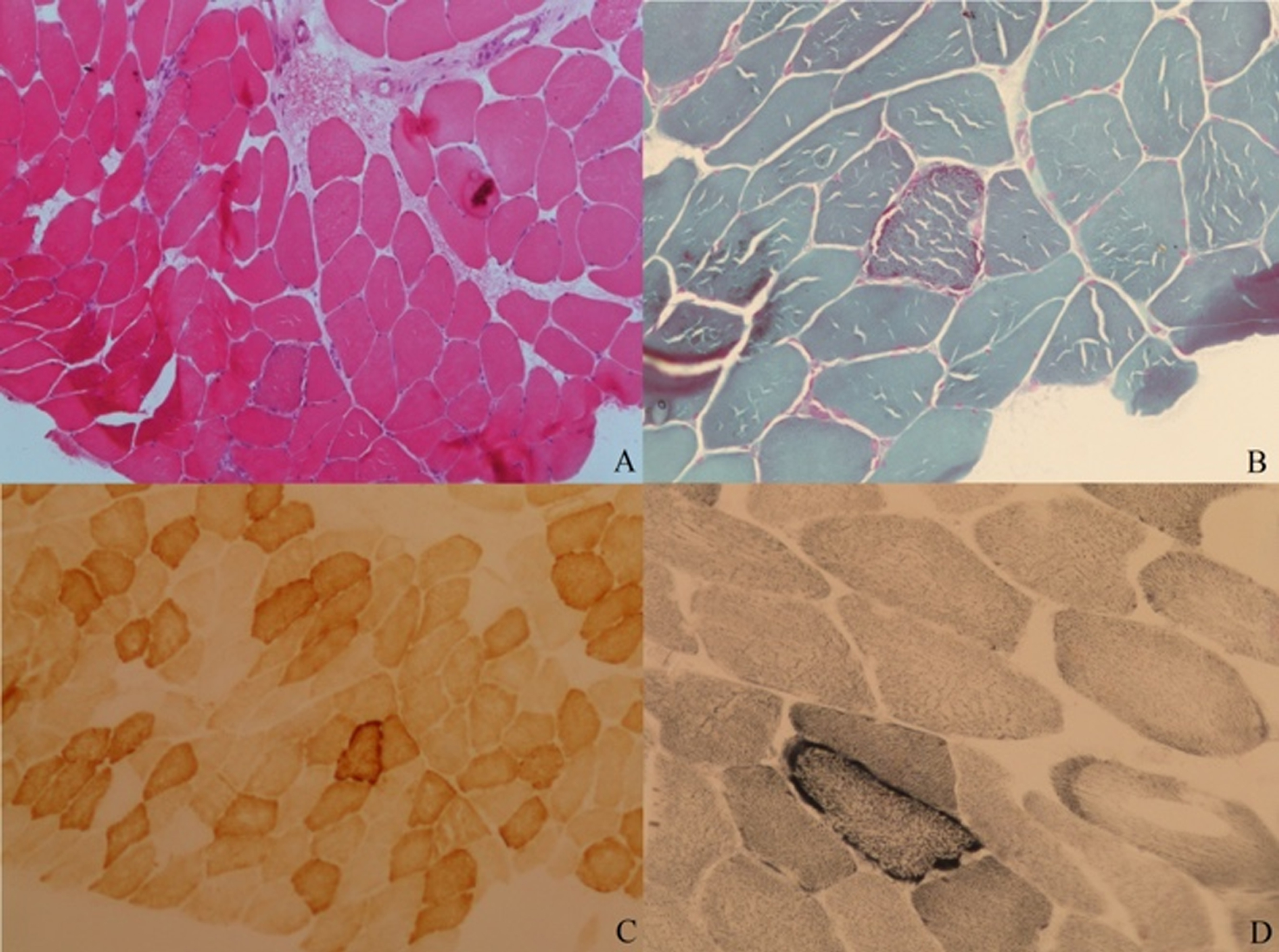
Muscle biopsy (original magnification 10X (A and C) and 20X (B and D)): (A) hematoxylin eosin; (B) Gomori trichrome stain.; (C) staining for cytochrome oxidase; (D) succinate dehydrogenase (SDH) staining.

Long-PCR analysis of mitochondrial DNA in muscle tissue: on the left, a normal control (number 7); on the right (number 8), the test performed on our patient, which shows a single deletion of mtDNA from 7061bp to 13613bp.
Accordingly, western blot analysis of lamin A/C in patient skeletal muscle showed the expected lamin A and lamin C bands and an additional band (around 76 kDa) (Fig. 5). The upper band corresponded to mutated lamin A, as expected from the mutation analysis and from the absence of any corresponding band in control or other EDMD2 muscle sample. The whole amount of A type lamins was reduced in EDMD2 skeletal muscle, although the patient carrying both LMNA and mtDNA defect showed a less pronounced reduction of protein levels.
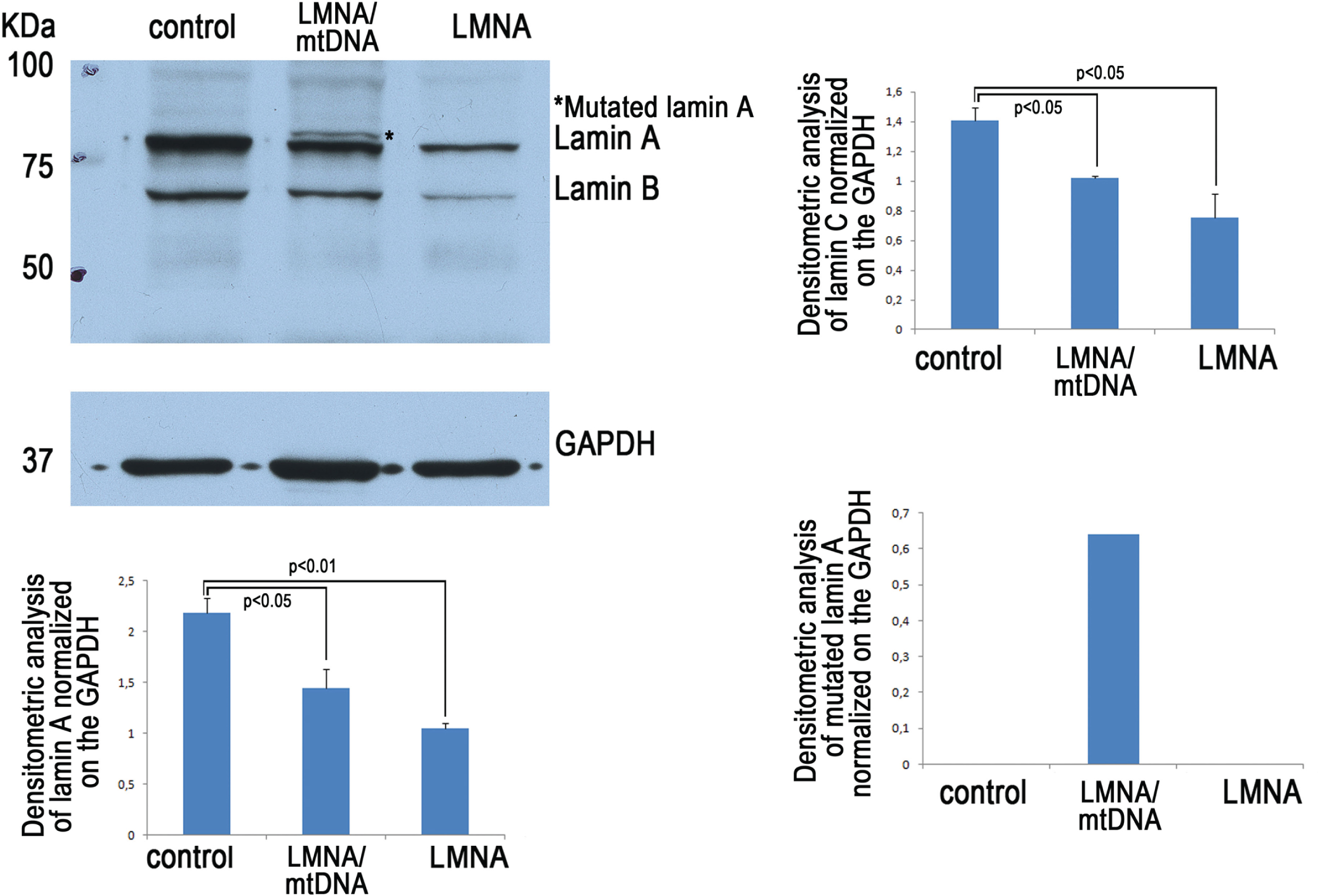
Western blot analysis of lamin A/C in biopsies of control skeletal muscle (control, an healthy subject undergoing orthopedic surgery for trauma), EDMD2-mtDNA patient (LMNA/mtDNA) and a “classical” EDMD2 patient (LMNA). GAPDH was used as a loading control and densitometric values were normalized to GAPDH. Data reported in the graphs are means of densitometric values obtained in three independent analyses. Statistically significant differences (p < 0.05 or p < 0.01) are indicated.
We therefore decided to better evaluate the possible associated cardiac involvement. The patient performed cardiac MRI, showing a small late enhancement area in the interventricular septum (Fig. 6), as reported in LMNA related cardiomyopathies [5]. Moreover, a first-degree atrioventricular block (PR of 300 msec) was detected. An incremental aerobic exercise test with cycle ergometer showed an anaerobic threshold reached at 39%of maximal oxygen uptake (%VO2 max).
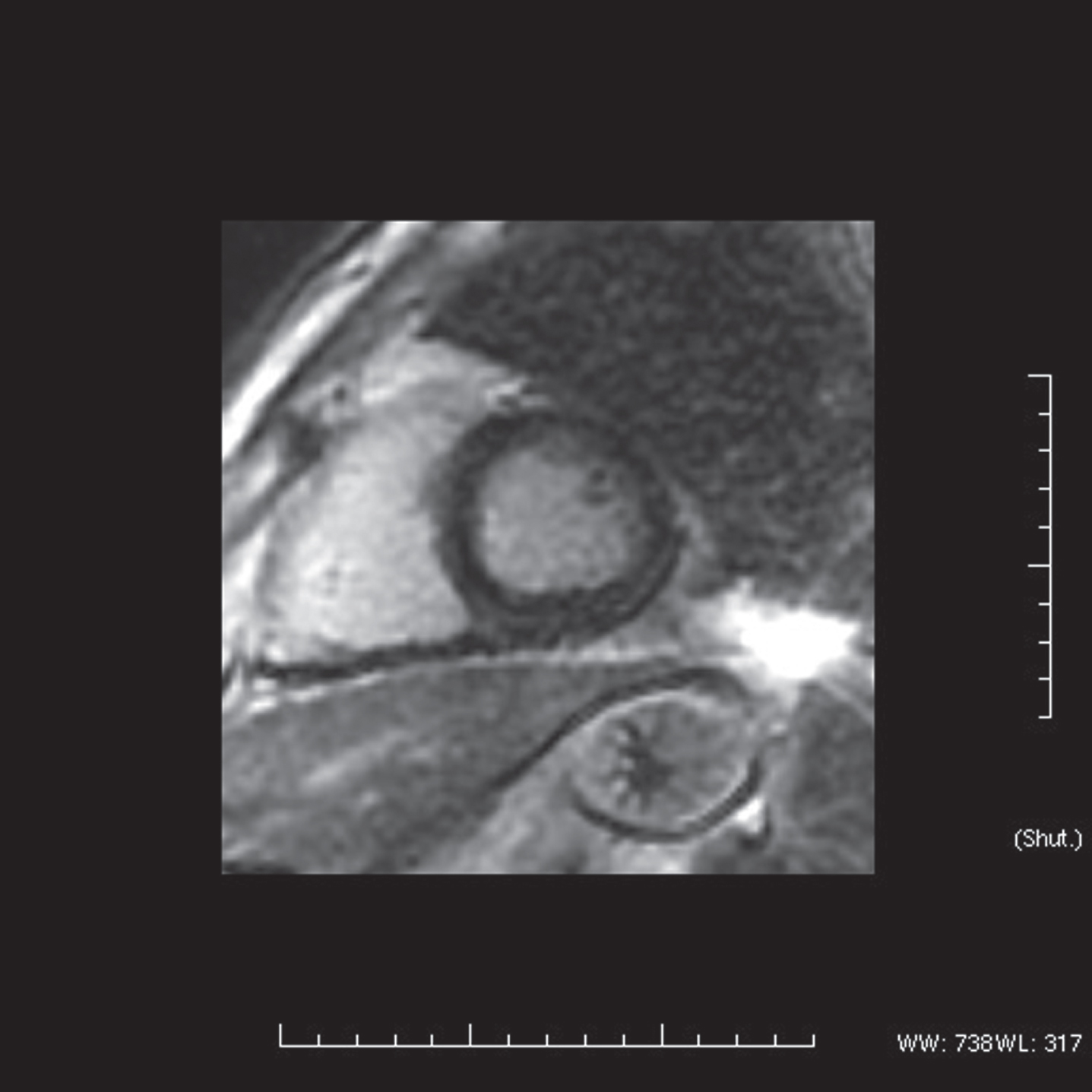
Cardiac MRI (late gadolinium enhancement sequence) showing a small area of midwall myocardial fibrosis in the basal inferior interventricular septum.
The segregation analysis was then performed in the available relatives, including son, sister and niece, all resulting carriers of the same LMNA variant; cardiological assessment in these subjects is ongoing.
DISCUSSION
A common pattern of muscle weakness in myopathies is symmetric weakness affecting predominantly the proximal muscles of the legs and arms, or the so-called “limb-girdle” distribution. LGMD is the fourth most common muscular dystrophy, but a broad range of genetic and acquired neuromuscular conditions share a similar presentation, including primary mitochondrial myopathies (both mitochondrial tRNA and nuclear genes mutations) [6]. New molecular technologies (next generation sequencing (NGS) and whole exome sequencing (WES) often show mutations in other genes besides the classic LGMD-associated genes, new genotype-genotype correlations and variants in multiple genes [7]. In these cases, muscle biopsy, clinical examination and other data are essential for the interpretation of molecular result. The presence of concomitant mutations in different genes may help to explain in part the phenotypic heterogeneity, some of the atypical presentations and complex phenotypes found in some patients, like in our case.
From a clinical point of view, the patient displayed a pattern of muscle involvement reminiscent of LGMD, characterized by proximal weakness and wasting especially of lower limb.
The co-occurrence of a mitochondrial DNA single deletion, which is a sporadic disorder, cannot be excluded. Muscular MRI assessment documented predominant involvement of the lower leg, with marked affection of the gastrocnemius medialis compared to the gastrocnemius lateralis, and at thigh of vasti and adductor, a pattern of involvement which confirmed LGMD1B [8]. Also, cardiac MRI showed interstitial myocardial fibrosis (detected by late gadolinium enhancement), which represents a subclinical marker of cardiac involvement in LMNA mutation carriers; interestingly, this finding in our patient was associated with a first-degree AV block, more common in carriers with interstitial fibrosis, as recently described (5). LMNA-related diseases have a variable age of onset, sometimes in late adulthood, as in our case. An earlier presentation is usually associated with marked cardiac involvement, with pacemaker implantation at a younger age and atrio-ventricular (A-V) block or rhythm disturbances, usually occurring before evidence of cardiomyopathy. Western blot analysis detected a reduced amount of LMNA/C, supporting pathogenicity of the new identified variant, as well as in silico prediction analysis. On the contrary, muscle biopsy showed prominent signs of mitochondrial involvement, which prompted mitochondrial DNA analysis. Basal lactate elevation and the anaerobic lactate threshold reduction also supported a mitochondrial alteration (40%of the normal maximal power output is reported to correspond to anaerobic lactate threshold in mitochondrial disorders).
Therefore, we can assume the following diagnostic hypothesis: (i) a double trouble, with single mtDNA deletion and LGMD1B coexisting in our patient; (ii) the patients is affected by LGMD1B with secondary mitochondrial involvement; (iii) co-occurrence of LGMD1B and age-related mitochondrial changes.
Mitochondrial abnormalities of ageing are characterized by the accumulation of COX negative fibers and mitochondrial DNA deletions. The common deletion was shown to be present in a wide variety of aged tissues including skeletal muscle. However, the deletion levels rarely exceed 1%, even in very old individual [9]. In our case, heteroplasmy was 15%and the size of the deletion was not of the common mtDNA deletion. Mitochondrial alteration, like swollen mitochondria, altered mitochondrial dynamics and membrane potential, activation of mitochondrial pathways of apoptosis and enhanced oxidative stress has been described in mouse, drosophila and human fibroblast laminopathy models [10–13]. To our knowledge, secondary mitochondrial involvement with a single mtDNA deletion has never been described in laminopathies and, however, cannot be excluded in our case.
In conclusion, although secondary mitochondrial involvement can not be ruled out, our case could represent an example of “double trouble” syndrome, in which the co-occurrence of one condition can modulate the phenotype of the other. Notably, we discovered the same LMNA variants in asymptomatic relatives, although a phenotypic heterogeneity is well documented among carriers of LMNA mutations with many cases of cardiological involvement without muscle weakness.
The accurate diagnosis and recognition of patients with a double-trouble genetic disease is essential for genetic risk assessment, management and prognosis, and potential therapeutic interventions.
Footnotes
ACKNOWLEDGMENTS
The authors are grateful to the following supporting and funding subjects: European Reference Network for Neuromuscular Diseases (ERN EURO-NMD), AIFA (TREAT LMNA project), Telethon-MITOCON grant GSP16001. MM was partially supported by RF-2016-02361495 and the EJPRD2019 project GENOMIT. The funder had no role in study design, writing of the report, or data collection, analysis, or interpretation.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest regarding the publication of this paper.