
Abstract
Background:
X-linked myotubular myopathy (XLMTM) is a life-threatening congenital myopathy that, in most cases, is characterized by profound muscle weakness, respiratory failure, need for mechanical ventilation and gastrostomy feeding, and early death.
Objective:
We aimed to characterize the neuromuscular, respiratory, and extramuscular burden of XLMTM in a prospective, longitudinal study.
Methods:
Thirty-four participants < 4 years old with XLMTM and receiving ventilator support enrolled in INCEPTUS, a prospective, multicenter, non-interventional study. Disease-related adverse events, respiratory and motor function, feeding, secretions, and quality of life were assessed.
Results:
During median (range) follow-up of 13.0 (0.5, 32.9) months, there were 3 deaths (aspiration pneumonia; cardiopulmonary failure; hepatic hemorrhage with peliosis) and 61 serious disease-related events in 20 (59%) participants, mostly respiratory (52 events, 18 participants). Most participants (80%) required permanent invasive ventilation (>16 hours/day); 20% required non-invasive support (6–16 hours/day). Median age at tracheostomy was 3.5 months (95% CI: 2.5, 9.0). Thirty-three participants (97%) required gastrostomy. Thirty-one (91%) participants had histories of hepatic disease and/or prospectively experienced related adverse events or laboratory or imaging abnormalities. CHOP INTEND scores ranged from 19–52 (mean: 35.1). Seven participants (21%) could sit unsupported for≥30 seconds (one later lost this ability); none could pull to stand or walk with or without support. These parameters remained static over time across the INCEPTUS cohort.
Conclusions:
INCEPTUS confirmed high medical impact, static respiratory, motor and feeding difficulties, and early death in boys with XLMTM. Hepatobiliary disease was identified as an under-recognized comorbidity. There are currently no approved disease-modifying treatments.
Keywords
INTRODUCTION
X-linked myotubular myopathy (XLMTM) is a life-threatening congenital myopathy caused by variants in the MTM1 gene, leading to absence or insufficiency of functional myotubularin protein. Myotubularin is a broadly expressed lipid phosphatase required for the normal development, maturation, and maintenance of skeletal muscle [1]. Natural history studies have shown that most patients with XLMTM typically experience profound muscle weakness from birth, significant respiratory insufficiency requiring chronic invasive mechanical ventilation, dysphagia requiring feeding support, absent or delayed major motor milestone attainment, and high rates of early mortality [2–6] most often related to respiratory failure [5, 7–9]. Approximately 20% of XLMTM patients have a milder phenotype in which they can walk independently and may live into adulthood, though most do ultimately require some form of ventilator support (often noninvasive) and feeding assistance [2, 11]. Historically, it has been estimated that approximately half of infants died before 18 months of age, and survival is highly dependent on intensive medical intervention [3, 6]. XLMTM has an estimated incidence of 1 in 40,000-50,000 newborn males [12].
There are currently no approved disease-modifying therapies for XLMTM, which is managed with multidisciplinary supportive care for manifestations of chronic, profound muscle weakness. Novel therapeutic strategies have shown promise in pre-clinical disease models, [13–16] including adeno-associated virus (AAV)-mediated gene replacement therapy [17–20].
The INCEPTUS study (NCT02704273) began in 2016 in preparation for ASPIRO (NCT03199469) —the first-in-human clinical study of an investigational gene replacement therapy in patients with XLMTM, which was anticipated to enroll young patients with a ventilator-dependent phenotype. INCEPTUS was designed to obtain longitudinal data on XLMTM natural history and prospectively characterize a control group for ASPIRO. Although prior natural history studies have characterized the clinical manifestations of XLMTM, a robust set of repeated longitudinal assessments of ventilatory dependence, respiratory muscle strength, motor milestones, and quality of life is a prerequisite for evaluating a novel disease-modifying therapy.
METHODS
Study design and participants
INCEPTUS was a prospective, non-interventional, multicenter study to evaluate patients < 4 years old at enrollment with genetically confirmed XLMTM and mechanical ventilatory support (invasive or noninvasive). Participant inclusion and exclusion criteria are shown in Table 1. Participants were evaluated every 3 months throughout the INCEPTUS study. For participants who continued into ASPIRO, INCEPTUS participation provided baseline measurements and control data prior to gene therapy administration.
Inclusion/exclusion criteria for the INCEPTUS Study
Inclusion/exclusion criteria for the INCEPTUS Study
BiPAP, bilevel positive airway pressure; CPAP, continuous positive airway pressure; XLMTM, X-linked myotubular myopathy.
INCEPTUS was conducted in accordance with International Conference on Harmonisation Good Clinical Practice, the Declaration of Helsinki, and Clinical Investigation of Medicinal Products in the Pediatric Population. Study protocols were approved by institutional review boards or ethics committees of each institution. Signed informed consent was obtained from legal guardians.
Assessments
Disease-related events, survival and growth
Disease-related adverse events and serious adverse events were recorded and coded using the Medical Dictionary for Regulatory Activities (MedDRA® version 20.0). Serial liver ultrasounds were performed to monitor for hepatic peliosis, a life-threatening complication affecting 5–10% of XLMTM patients [2, 8]. Liver function tests (LFTs) included alanine aminotransferase (ALT), aspartate aminotransferase (AST), total and direct bilirubin, and gamma-glutamyl transferase (GGT). Although cardiac disease has not been described as a significant feature of XLMTM, troponins T/I, creatine kinase isoenzymes, 12-lead electrocardiogram, and echocardiography were performed to establish baseline parameters. Anthropometrics and survival were also assessed.
Motor function
Motor function was assessed using the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND), [21, 22] validated previously in XLMTM patients [23].
Participants able to sit upright unassisted as assessed by the clinical evaluator or who scored > 45 points on CHOP INTEND (n = 7) also had motor function assessed using Bayley Scales of Infant and Toddler Development III (Bayley III) [24]. In addition, the Motor Function Measure-20 (MFM-20) validated in patients with neuromuscular disease, [25] was performed in participants > 2 years old. Assessments were video recorded to ensure correct administration of standardized procedures and enable ongoing quality control and training.
Respiratory function
Ventilator dependence was assessed as parent-reported amount of time on ventilator support in the preceding 24-hour period. Maximal inspiratory and expiratory pressures (MIP/MEP) were obtained in accordance with American Thoracic Society/European Respiratory Society recommendations for respiratory muscle testing [26].
Gastrostomy feeding support and management of bronchial secretions
The number of participants requiring a gastrostomy tube and time from birth to its placement were calculated. The Parental Global Impression of Secretion Severity (PGIS-S) [27] was used to assess daily secretion management (oral, nasal and bronchial) and suctioning requirements (see Supplementary Methods).
Health-related quality of life
Health-related quality of life and impact of disease on caregivers were evaluated using the Assessment of Caregiver Experience with Neuromuscular Disease (ACEND) [28] and the Pediatric Quality of Life Inventory Neuromuscular Module (PedsQL-NM) (see Supplementary Methods) [29, 30].
Statistical methods
A single-arm mixed-effect model with fixed effect of time was used to examine least square (LS) means (standard error [SE]) change from baseline in disease parameters. Median and 95% confidence interval (CI) were reported for longitudinal endpoints and mean with standard deviation (SD) for endpoints measured at a single time-point. Study duration was not fixed, resulting in different end of study timepoints for each participant, to allow eligible participants to enroll in the ASPIRO study.
RESULTS
Between August 2016 and June 2019, 34 children with XLMTM enrolled at eight sites in North America and Europe (Supplementary Table 1). Median (range) follow-up was 13.0 (0.5, 32.9) months, with 23 participants (68%) followed for at least 6 months and 18 participants (53%) for at least 12 months (Supplementary Figure 1). Baseline participant characteristics are shown in Table 2. Heights and weights were within the standard growth chart ranges, except for two participants with documented macrosomia.
Demographic and clinical characteristics at study enrollment
Missing baseline data imputed with first available post-baseline value. CHOP INTEND: Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders; ULN: upper limit of normal (of given reference range). *Age was calculated in years from date of birth to date of screening. Due to privacy laws in certain countries, full birth date cannot be reported. For these participants, date of birth is recorded as 01 Jan of the actual birth year, resulting in overestimate of age at enrollment in some participants.
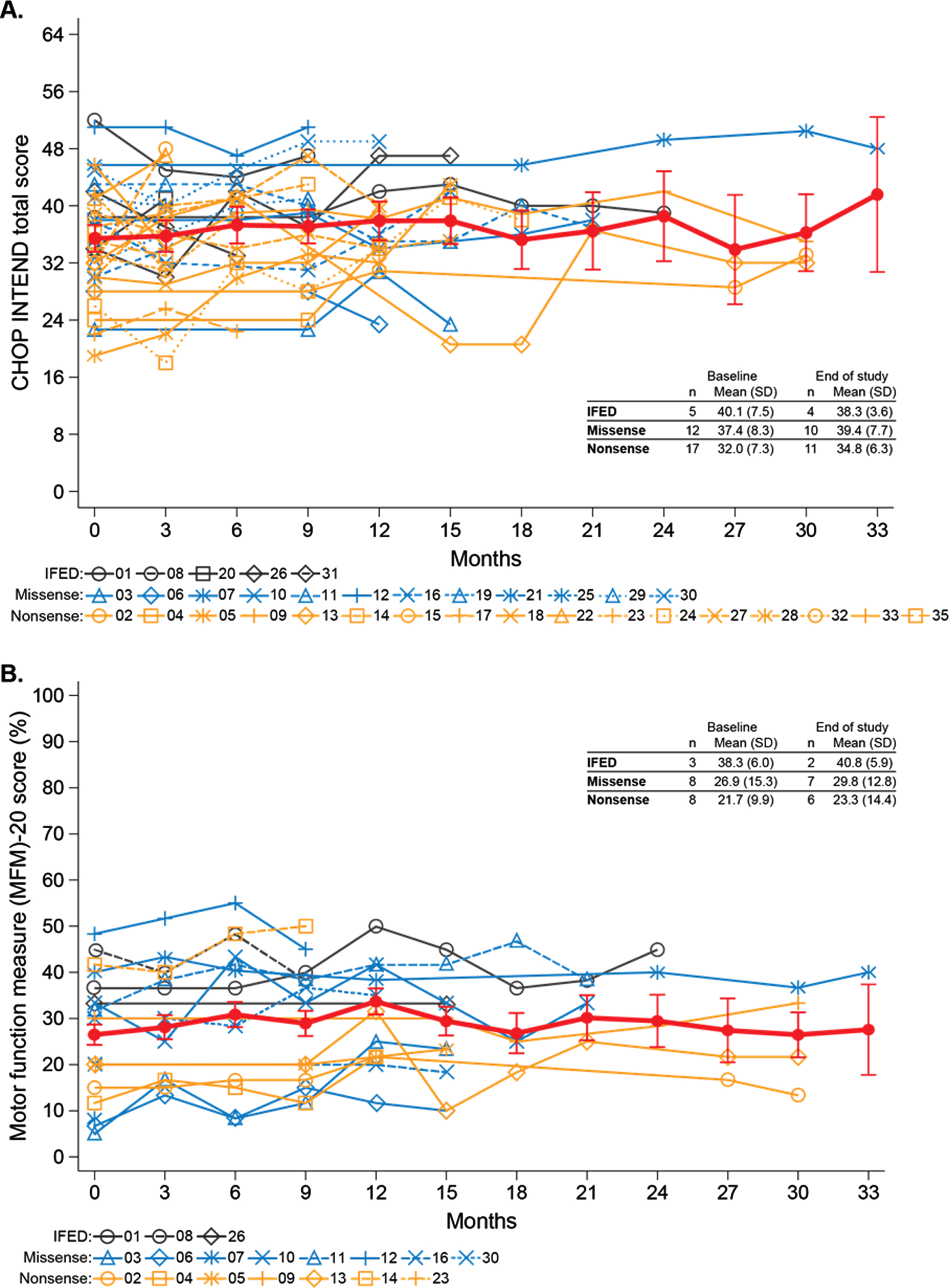
Motor function over time for individual participants by mutation type. (A) CHOP INTEND total scores. (B) MFM-20 scores. The thick red lines represent LS means and standard errors from a mixed-effect model with fixed effect of time.
Most participants had extensive histories of medical conditions and procedures related to XLMTM (Table 3). Half of participants had nonsense MTM1 mutations that would be expected to produce little or no functional or stable myotubularin, 35% had missense mutations and 15% in-frame exonic deletions (IFED), possibly expressing stable intact or internally deleted proteins with residual myotubularin activity (Supplementary Table 2).
Medical co-morbidities and procedures reported in≥20% of INCEPTUS participants at study enrollment
Disease-related events, survival, and histopathology
Overall, 61 disease-related serious adverse events were reported in 20 (59%) participants, primarily respiratory complications (85%) (Supplementary Table 3). Serious adverse events were associated with 52 hospitalizations among 18 participants, with a mean (SD) duration of 8.1 (7.6) days.
Three deaths occurred (participants 04 [3.5 years], 14 [3.5 years], and 15 [2.3 years]) associated with serious adverse events of cardiopulmonary failure, aspiration pneumonia, and hepatic hemorrhage with evidence of diffuse hepatic peliosis, respectively. In all three participants, histopathological findings were characteristic of XLMTM in skeletal muscles, including myofiber smallness, excessive internal nucleation, and organelle mislocalization (Supplementary Figure 2) [31]. These findings affected > 80% of fibers in the muscles sampled in two of the deceased participants (14 and 15). More extensive sampling of skeletal muscles in participant 04 showed a variable but moderate degree of pathology characteristic of XLMTM. None showed myofiber degeneration, necrosis, or fatty infiltration.
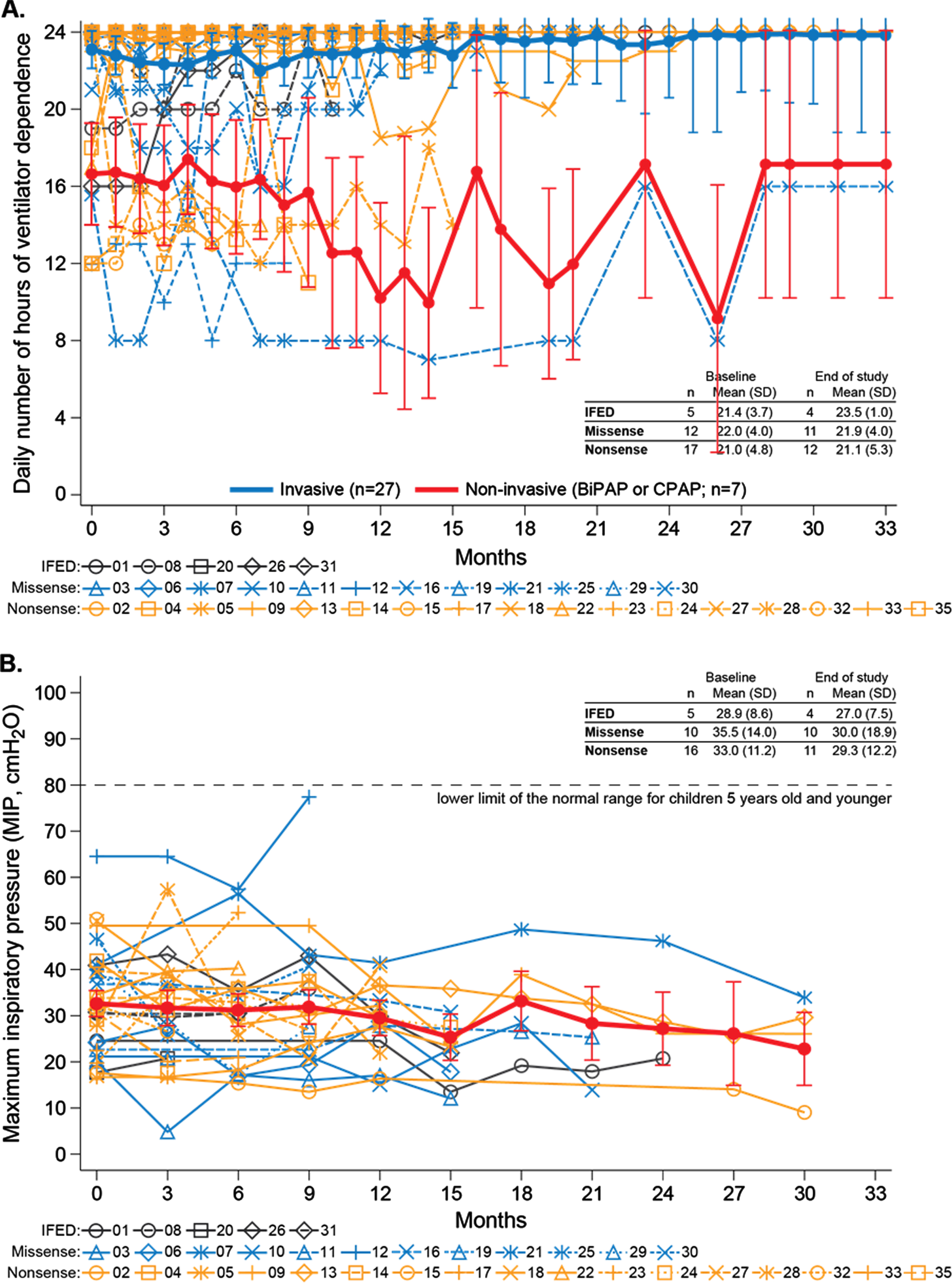
Respiratory function over time for individual participants by mutation type. (A) Ventilator dependence within the previous 24 hours. Higher ventilator dependence reflects more daily hours of ventilator use. The thick blue and red lines represent LS means and standard errors from a mixed-effect model with fixed effect of time for invasively ventilated participants (blue) and noninvasively ventilated participants (red). (B) Maximal inspiratory pressure (MIP). The thick red line represents LS means and standard errors from a mixed-effect model with fixed effect of time.
Hepatic disease
At enrollment, 8 (24%) participants had a history of hepatic disease, including continuous or intermittent cholestasis and hyperbilirubinemia (Table 4). All participants had at least one LFT during the study; 29 (85%) had at least one elevated value for ALT, 21 (62%) for AST, and 12 (35%) for total or direct bilirubin. Twenty-one (62%) participants had either hepatic adverse events or ultrasonographic imaging abnormalities or both, and 17 (50%) received medications commonly administered for cholestatic or other hepatic disease (Table 4). Overall, 91% of participants had a history of hepatobiliary disease at enrollment or showed at least one sign of hepatic disease during the prospective study period.
Adverse events, medications, and ultrasonographic and laboratory findings potentially related to liver disease among INCEPTUS participants
Color Legend: f1x ULN; 1.1-3x ULN; 3.1-5x ULN; 5.1-10x ULN;>10x ULN. ALT: alanine aminotransferase; AST: aspartate aminotransferase; GGT: gamma-glutamyl transferase; LFT: liver function test; ULN: upper limit of normal. *4 evaluable direct bilirubin values. †1 evaluable GGT value. — Denotes none.
Elevated LFTs and signs of hepatobiliary disease were present in participants across all three mutation types. Participants with nonsense mutations had the most prevalent and pronounced abnormalities, including 35% with at least one elevated direct bilirubin level, 88% with at least one elevated ALT, and 71% with at least one elevated AST (Table 4). Six of 17 (35%) participants with nonsense mutations had imaging findings consistent with hepatomegaly or hyper-echogenicity.
One participant death at age 2.3 years (participant 15) was attributed to liver hemorrhage with evidence of diffuse hepatic peliosis and liver histopathology showing numerous areas with blood pools of varying size not lined by endothelial cells (consistent with hepatic peliosis), and rare focal areas of necrosis without evidence of significant inflammation, fibrosis, or cholestasis. In participant 04, who died at age 3.5 years, postmortem liver examination revealed hepatomegaly (58% greater weight than age-appropriate comparator) without evidence of cholestasis, fibrosis, or other lesions.
Motor function
Participants completed CHOP INTEND, Bayley III, and MFM-20 assessments. The mean [SD] CHOP INTEND score was virtually unchanged from baseline (35.1 [8.1]) to end of study (37.2 [6.7]) (Fig. 1A). These scores reflect severely limited motor function (healthy children typically achieve the maximum score of 64 by 3–6 months of age [23]), and individual item scores show that participants failed to score age-appropriately on essential items that are prerequisites for achieving major motor milestones (Supplementary Figure 3). The mean [SD] MFM-20 total score was also virtually unchanged from baseline (26.5% [12.9% ]) to end of study (28.7% [13.5% ]) (standardized to 100% scale [25]), reflecting static but impaired motor function over time (Fig. 1B). CHOP INTEND and MFM-20 scores remained static across all mutation groups over time (Fig. 1A-B).
For Bayley III assessments, median (interquartile range) participant age was 2.6 years (1.9, 3.0) and 2.9 years (2.1, 3.9) at first and last assessments, respectively. Seven participants (21%) could sit unsupported for ≥30 seconds, but one lost this ability during the study. Two of these participants had IFED, two had missense mutations, and three had nonsense mutations, including the participant who lost the ability to sit unsupported. No participants achieved higher level motor milestones such as pulling to stand, standing, or walking with or without support.
Respiratory function
Approximately 80% of participants required a tracheostomy (n = 27). Median time from birth to tracheostomy was 3.5 months (95% CI, 2.5, 9.0). Mean [SD] daily ventilation hours remained constant from baseline (21.4 [4.3]) to end of study (21.7 [4.4]), regardless of ventilation type and mutation type. Mean ventilator dependence was similar across mutation groups at baseline and remained high over time (Fig. 2A and Supplementary Table 4). Participants requiring invasive ventilation did not improve to enable switching to non-invasive ventilation nor did those requiring non-invasive ventilation switch to invasive ventilation.
MIP was consistently well below normal levels (80 cmH2O) for age-matched children [32] (Fig. 2B). Mean [SD] MIP decreased slightly from baseline (33.2 [11.7] cmH2O) to end of study (29.2 [14.3] cmH2O) (Fig. 2B), with similar decrements across all three mutation groups. There was no change in mean [SD] MEP from baseline (24.6 [12.5] cmH2O) to end of study (25.9 [12.0] cmH2O).
Ninety-seven percent (n = 33) of participants required feeding support via gastrostomy tube. Median time from birth to gastrostomy was 2.3 months (95% CI, 1.4, 3.2). Secretion management reported by PGIS-S was impaired in almost all participants at baseline, and mean [SD] PGIS-S score was unchanged from baseline (3.9 [1.3]) to end of study (3.9 [1.1]) (Supplementary Figure 4).
Caregiver experience and health-related quality of life
At baseline, ACEND total scores were between 13 and 63 (Supplementary Figure 5A) standardized to a scale of 0% (full caregiver support required) to 100% (no support required). The mean [SD] ACEND total score was virtually unchanged from baseline (31.8 (11.9]) to end of study (29.0 [12.8]). The mean [SD] PedsQL-NM total score decreased slightly from baseline (50.3 [13.8]) to month 30 (44.3 [15.1]) (Supplementary Figure 5B), on a scale from 0 (lowest QoL) to 100 (highest QoL).
DISCUSSION
INCEPTUS is the most comprehensive study thus far to quantify the extensive disease burden of XLMTM prospectively and longitudinally, including the persistence over time of ventilator dependence, low respiratory muscle strength, absent or severely delayed motor milestones, and diminished quality of life, in young boys with ventilator-dependent disease. These longitudinal data from 34 participants with XLMTM and ventilator dependency expand upon previous natural history studies showing severely compromised respiratory and motor function that fails to improve over time [2, 7]. In INCEPTUS, quarterly evaluations were performed to quantify the most comprehensive array of disease parameters to date. All INCEPTUS participants were permanently dependent on ventilator support, most were unable to achieve motor milestones, and most suffered severe medical events. Our data show that, although ventilator support extends the lifespan, respiratory and motor function impairments remain static.
Despite receiving high-quality, specialized, clinical care during INCEPTUS, participants had minimal to no improvement on any parameters and nearly 10% died from disease-associated complications. This unmet medical need for children with XLMTM is further underscored by the extensive care requirements demonstrated by ACEND scores, including constant monitoring by caregivers, multiple extended hospitalizations, and the need for frequent suctioning of secretions by caregivers, which also did not improve over the course of the study.
Patients requiring invasive ventilation for > 16 hours per day (80% in INCEPTUS) are unlikely to have intrinsic capacity to wean from ventilatory support [33]. Pediatric patients on mechanical ventilation due to a variety of underlying diagnoses have increased risk of mortality, [34] often from causes other than progression of the initial diagnosis, [35] and substantial physical, emotional, and financial demands associated with reliance on mechanical ventilation [36–38]. The importance of reducing ventilator dependence is highlighted by 85% of serious adverse events in INCEPTUS being respiratory in nature. Similar observations in a one-year longitudinal study demonstrated ventilator support time as a reliable, reproducible, robust, and clinically meaningful measure of disability in XLMTM patients [2].
Healthy infants typically reach the maximum CHOP INTEND score of 64 in the first 3 to 6 months of life [39] and can usually roll from back to belly and sit unassisted for > 30 seconds by 8 months of age [40]. None of the INCEPTUS participants achieved the maximum CHOP INTEND score despite being older than 6 months. CHOP INTEND score trajectories were essentially flat, highlighting a lack of expected progression in motor function in early life. Bayley-III data showed that 79% of participants were unable to sit unsupported for≥30 seconds. None achieved higher level motor skills, such as pulling to stand or walking with or without assistance, despite some being over 6 years old at their last evaluation.
Recent reports demonstrate hepatobiliary disease associated with the natural history of XLMTM (i.e., not associated with any specific treatments). A recent case series highlighted five untreated XLMTM patients with recurrent cholestasis, including one who has progressed to end-stage liver failure [41]. Furthermore, a retrospective analysis of hepatic data in a cohort of Italian patients with XLMTM indicates the presence of cholestasis [42]. Additional cases include a child with hypertransaminemia and hyperbilirubinemia (up to 40 times ULN) with hepatopathy and cholestasis [43] and another child with hyperbilirubinemia and elevated urine bile acids consistent with asymptomatic cholestasis leading to severe vitamin K deficiency, coagulopathy and fatal intracranial hemorrhage [44]. One-quarter of INCEPTUS participants had a history of hepatobiliary disease prior to enrollment; during prospective monitoring, 91% experienced hepatobiliary disease or hepatic adverse events or laboratory or ultrasonographic abnormalities (Table 4). Although serum bile acid levels were not measured in INCEPTUS, observed elevated transaminase and bilirubin levels and ultrasonographic abnormalities may have been reflective of underlying cholestasis. As there was not high awareness of intrahepatic cholestasis in XLMTM patients at the time of INCEPTUS, there was no determination of underlying reasons for isolated AST or ALT elevations (i.e., liver damage, infection, or transient intermittent increase). However, the extent of aminotransferase elevations, as well as the relative elevations of ALT compared to AST, are suggestive of a hepatogenic source. Based on emerging reports of intrahepatic cholestasis complicating XLMTM, [41–44] in combination with the high frequency of liver-related laboratory abnormalities described in this study, we postulate that monitoring of liver parameters (including potential consultation with a pediatric hepatologist) should be an important aspect of clinical care moving forward. Obtaining liver-related laboratory parameters (ALT, AST, GGT, total and direct bilirubin), plus evaluation of serum bile acid levels, may help identify cholestatic episodes in XLMTM patients, and will also enhance understanding of the incidence of this phenotype in the XLMTM population and the potential triggers for its occurrence. Liver biopsy is a useful modality for the diagnosis of cholestasis and for better defining its cause but is potentially contraindicated in XLMTM due to the risk of hepatic peliosis. Unfortunately, imaging modalities (such as ultrasound or MRI) have not demonstrated great utility for the diagnosis of intrahepatic cholestasis and the determination of its causes. Histopathologic evaluation for intrahepatic cholestasis and secondary injury was not performed, as liver biopsy is relatively contraindicated in XLMTM patients due to potential coexistent peliosis and subsequent risk for life-threatening hemorrhage [45]. Myotubularin may play a role in cellular processes outside of muscle cells, including intracellular/endosomal trafficking and bile export from hepatocytes [46]. Further investigation of myotubularin’s role in hepatobiliary function is needed. Preliminary experience with AAV-mediated gene replacement therapy in the first-in-human ASPIRO study includes serious hepatotoxicity leading to the death of 4 participants. Amongst surviving patients, clinically meaningful improvements in muscle strength and function were observed [47]. Currently, the study is on clinical hold until a benefit-risk assessment of proceeding with this therapy can be determined. Treated participants continue to be monitored and assessed per study protocol.
Despite the broad range of ages at enrollment in INCEPTUS (0.2 to 3.4 years), participants from all mutation groups had profound impairment and absence of improvement in disease parameters. Of note, patients with nonsense mutations tended to have more pronounced signs of hepatobiliary disease (Table 4), and non-truncating mutations have been associated with less pronounced decrements in respiratory function compared with truncating mutations [5].
INCEPTUS participation was restricted to patients who required some form of ventilator support at baseline, excluding a subset of patients who may have milder disease [2]. Nevertheless, XLMTM is widely recognized as a severe disease in which the vast majority of patients require ventilator support, and the INCEPTUS population was comparable to cohorts from previous natural history studies [2, 7]. It is also important to note that, while the median follow-up in INCEPTUS was 13 months and 53% of participants had at least 12 months’ follow-up, one-third of participants had less than 6 months follow-up. A limitation of INCEPTUS is that, at the time of the study, there was not good general awareness of the potential for intrahepatic cholestatic disease in XLMTM patients. Thus, we did not collect data, such as serial serum bile acid levels, that would have been ideal for evaluating this.
CONCLUSIONS
Children with ventilator-dependent XLMTM have profoundly impaired respiratory and motor function that do not spontaneously improve and experience frequent medical complications leading to hospitalization and fatal events. Evidence of underlying hepatobiliary disease, present in almost all INCEPTUS participants, may represent an underappreciated aspect of XLMTM pathophysiology. In the INCEPTUS cohort, the high ventilator dependence, low respiratory capacity, and severely limited motor function remained static throughout the observation period.
Footnotes
ACKNOWLEDGMENTS
The INCEPTUS and ASPIRO studies are sponsored by Astellas Gene Therapies. The authors thank the children and families with XLMTM who allowed their data to be collected for the INCEPTUS and ASPIRO studies, and the entire XLMTM patient community for their cooperation and participation in the development of the studies, including: the Joshua Frase Foundation, MTM-CNM Family Connection, The Myotubular Trust, Where There’s a Will There’s A Cure Foundation for Myotubular Myopathy, and ZNM –Zusammen Stark! Clinical site management and regulatory support was provided by Pharm-Olam, Inc. We would like to thank the clinical team; Bhargav Achanta and, Aparna Shetty, PhD from Astellas Gene Therapies for critical contributions to publication development. We thank Laurie LaRusso of Chestnut Medical Communications for medical writing support funded by Astellas Gene Therapies.
CONFLICTS OF INTEREST
JJD reports research grants from Astellas Gene Therapies,* serving on advisory boards for Dynacure, Kate Therapeutics, and RYR1 Foundation, and serving as an editor of the Journal of Neuromuscular Diseases. BKS reports consulting fees from Astellas Gene Therapies* and Sarepta. CGB is editor-in-chief of the Journal of Neuromuscular Diseases. DAB reports former participation on the Scientific and Clinical Advisory Board for Astellas Gene Therapies* and receiving travel expenses for attending advisory board meetings. AB reports grants or personal fees from Biogene and Sanofi Genzyme; advisory or other board participation at Dynacure and CMD Scientific and Medical. LL reports consulting fees paid to her institution for training of evaluators in the INCEPTUS study. MWL reports research grants paid to his institution from Astellas Gene Therapies,* Solid Biosciences, Kate Therapeutics, Taysha Therapeutics, and Prothelia and consulting fees from Astellas Gene Therapies,* Encoded Therapeutics, Modis Therapeutics, Lacerta Therapeutics, AGADA Biosciences, Dynacure, Affinia, and Biomarin. TD reports personal consulting fees from Astellas Gene Therapies,* Biogen, Avexis, Roche, Novartis, Dynacure, Sarepta, Pfizer, and Genentech. GR reports consulting fees from Astellas Gene Therapies* paid to his institution. VM reports consulting fees from Astellas Gene Therapies* paid both directly to her and her institution. PBS reports grants or personal fees from Astellas Gene Therapies,* Sarepta, AveXis, PTC Therapeutics, Pfizer, Biogen, Argenx, Catalyst, Roche, Ra Pharma, Grifols, Alexion, CSL Behring, Fulcrum Therapeutics, Fibrogen, Acceleron, Reveragen, Sanofi, and Santhera. AHB reports sponsored research support from NIH, MDA (USA), AFM Telethon, Alexion Pharmaceuticals Inc., Astellas Gene Therapies,* Dynacure SAS, and Pfizer Inc. He has consulted and received compensation or honoraria from Astellas Gene Therapies,* Biogen, F. Hoffman-La Roche AG, Kate Therapeutics, GLG Inc, Guidepoint Global, and Novartis, holds equity in Kate Therapeutics and Kinea Bio, and is an inventor on a US patent describing a method for gene therapy of XLMTM. LNA reports grants or contracts from Astellas Gene Therapies* for clinical evaluator training and quality control in the INCEPTUS and ASPIRO studies. RJG reports limited consulting fees from Astellas Gene Therapies* for work on the ASPIRO study design and advisory board participation for Astellas Gene Therapies,* Biogen, and Genetech. CL reports consulting fees, payments or honoraria, and/or travel expenses from Roche, Biogen, ATOM, AFEHM, Sysnav, and PeerVoice. LS reports consulting fees from Astellas Gene Therapies* and Dynacure. NLK reports advisory board participation for Astellas Gene Therapies,* Biogen, Genentech, Novartis and Sarepta. AMS, ET, MJ, FM, SN, TP, and WMF report no conflicts of interest. JC, MN, WS, and BS are employees and/or stockholders of Astellas Gene Therapies.* JL, CF, SC, ES, MCV, SP, and SR are former employees and/or stockholders of Astellas Gene Therapies.*
*Formerly Audentes Therapeutics