
Abstract
Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD) phenotypes are used to describe disease progression in affected individuals. However, considerable heterogeneity has been observed across and within these two phenotypes, suggesting a spectrum of severity rather than distinct conditions. Characterizing the phenotypes and subphenotypes aids researchers in the design of clinical studies and clinicians in providing anticipatory guidance to affected individuals and their families. Using data from the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet), we used K-means cluster analysis to group phenotypically similar males with pediatric-onset dystrophinopathy. We identified four dystrophinopathy clusters: Classical BMD, Classical DMD, late ambulatory DMD, and severe DMD. The clusters that we identified align with both ‘classical’ and ‘non-classical’ dystrophinopathy described in the literature. Individuals with dystrophinopathies have heterogenous clinical presentations that cluster into phenotypically similar groups. Use of clinically-derived phenotyping may provide a clearer understanding of disease trajectories, reduce variability in study results, and prevent exclusion of certain cohorts from analysis. Findings from studying subphenotypes may ultimately improve our ability to predict disease progression.
INTRODUCTION
Dystrophinopathies are X-linked muscular dystrophies resulting from mutations in the DMD gene. Historically, dystrophinopathies have been classified into two major phenotypes, Duchenne (DMD) or Becker (BMD) muscular dystrophy. DMD is a severe, progressive, and life-limiting disorder that affects skeletal, respiratory, and cardiac muscles; deficits in cognitive functions have also been reported. BMD is milder with slower disease progression, later or no loss of ambulation, and variable involvement of the cardiac and respiratory systems [1].
Considerable heterogeneity has been observed within the DMD and BMD phenotypes suggesting a spectrum of severity rather than two distinct conditions [2–5]. This phenotypic heterogeneity complicates the design and analysis of clinical research studies and prediction of disease progression. In an effort to group individuals with similar clinical presentations, investigators in previous studies defined potential DMD or BMD subphenotypes by identifying either distinct clusters or by describing a range of clinical presentations that do not necessarily represent classical definitions [3, 6]. Characterizing the phenotypes and subphenotypes can aid researchers in the design of clinical studies and clinicians in providing anticipatory guidance to affected individuals and their families.
The purpose of this analysis is to identify subphenotypes in a large, population-based cohort with clinically confirmed pediatric-onset dystrophinopathy and longitudinal clinical follow-up.
MATERIALS AND METHODS
Study population
The Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet) is a population-based surveillance system that collects demographic and clinical data on individuals with muscular dystrophy. Data from Arizona, Colorado, Georgia, Hawaii, Iowa, and western New York were used for this study [7]. Eligible individuals included those born on or after January 1, 1982, who were identified as having a dystrophinopathy diagnosis before 21 years of age and were residents of an MD STARnet site. Clinical and vital records information were collected from January 1, 1982 through December 31, 2011 or December 31, 2012 for those diagnosed in 2011. The case-finding methodology used by MD STARnet is based on active review of source records from neuromuscular clinics, hospital discharge databases, private physician practices, service sites for children with special health care needs, and birth defect surveillance programs [7, 8].
MD STARnet used an algorithm to classify all individuals in the dataset as DMD, BMD, or not classifiable according to three index variables: mobility, molecular, and onset [9]. The mobility index took into account ambulation beyond 16 years of age, ceased ambulating prior to age 12 years, or ceased ambulating prior to 16 years if steroid treatment was documented continuously for a minimum of two years prior to ambulation ceasing. The molecular index included either in-frame/out-of-frame mutation determination or presence/absence of dystrophin in western blot. All individuals included had molecular confirmation through either DNA testing or muscle biopsy dystrophin quantification. The onset index classified individuals based on age of onset prior to the 5th birthday and after the 10th birthday. This current analysis included those individuals in MD STARnet who had verified values for all three index variables.
Classification and clinical variables
We used the following clinical events to create the clusters as they were available for all cases in this dataset: 1) last age in years the individual was documented to be ambulatory, 2) age in years of symptom onset, and 3) age in years at last clinic visit (last age the individual was known to be living). Additionally, we included the following clinical variables related to severity of disease progression to further characterize the derived clusters: onset of cardiomyopathy (CM), initial noninvasive ventilation (NIV) utilization, and corticosteroid treatment. These clinical variables were not used to create the clusters but were used to determine the differences in overall severity in clinically relevant comorbidities across the clusters.
Ages were calculated in years using date of birth and corresponding event dates, with partial years to the second decimal point. Ages at last ambulation and last clinic visit consisted of latest documented ages still ambulating and age last documented as living (age at last clinic visit) or age at death, respectively. Symptom onset was the earliest age of documented symptoms in a medical chart specific to gait and mobility, including Gowers’ maneuver, trouble walking/running/climbing stairs, abnormal gait, falling behind peers in mobility, and gross motor delays [10]. Onset of CM was defined as the first age when echocardiographic shortening fraction fell below 28% or, if shortening fraction was unavailable, the first date when ejection fraction fell below 55%. Initiation of NIV was the earliest age at which Bilevel Positive Airway Pressure (BiPAP) or Continuous Positive Airway Pressure (CPAP) was noted in a clinical record. Periods of corticosteroid use were determined from dates of use entered into the abstraction database with discontinuation of use of at least one month to designate periods of nonuse.
Statistical analysis
K-means cluster analysis was used to determine clusters, and the Welch robust test for equality of means was used when data were non-normally distributed [11]. Due to variability in age at last clinic visit, Cox proportional hazards modeling with corticosteroid use as a time-dependent covariate was used to examine cluster differences on the clinical variables. Cases missing any of the three index variables were not included in the analyses. We further assessed the differing severity levels identified in the four clusters using common clinical outcomes, specifically age of CM onset and first use of NIV. Cox proportional hazards with steroid use as a time-dependent variable was used to compare clusters on NIV start and CM onset clinical outcomes. Lastly, we compared the clusters on presence of specific genetic mutations using chi-square. SAS version 9.4 © (Cary, NC) and SPSS version 24 © (IBM Analytics) were used to conduct the statistical analysis. Two independent analysts replicated the analyses and confirmed the results.
RESULTS
Of 1054 MD STARnet cases, 268 were verified to have all three index variables (Supplemental Figure 1). The 268 cases represent 232 individuals with a DMD MD STARnet assigned phenotype, 16 individuals with a MD STARnet assigned BMD phenotype, and 20 individuals who had conflicting index variables and were therefore flagged as Not Classifiable. Since the number of clusters cannot be known a priori, we selected two methods (Hierarchical agglomeration schedule and Euclidian distance) to estimate the ideal cluster number, which was between four and five. Visual review of the cluster plots (elbow) demonstrated a stronger visible demarcation of four clusters; therefore, four was selected as the final cluster count (Fig. 1). Although clusters were not all similar in size, they were spherical in nature and the different sizes of the clusters were in keeping with the prevalence differences between BMD and DMD phenotypes.
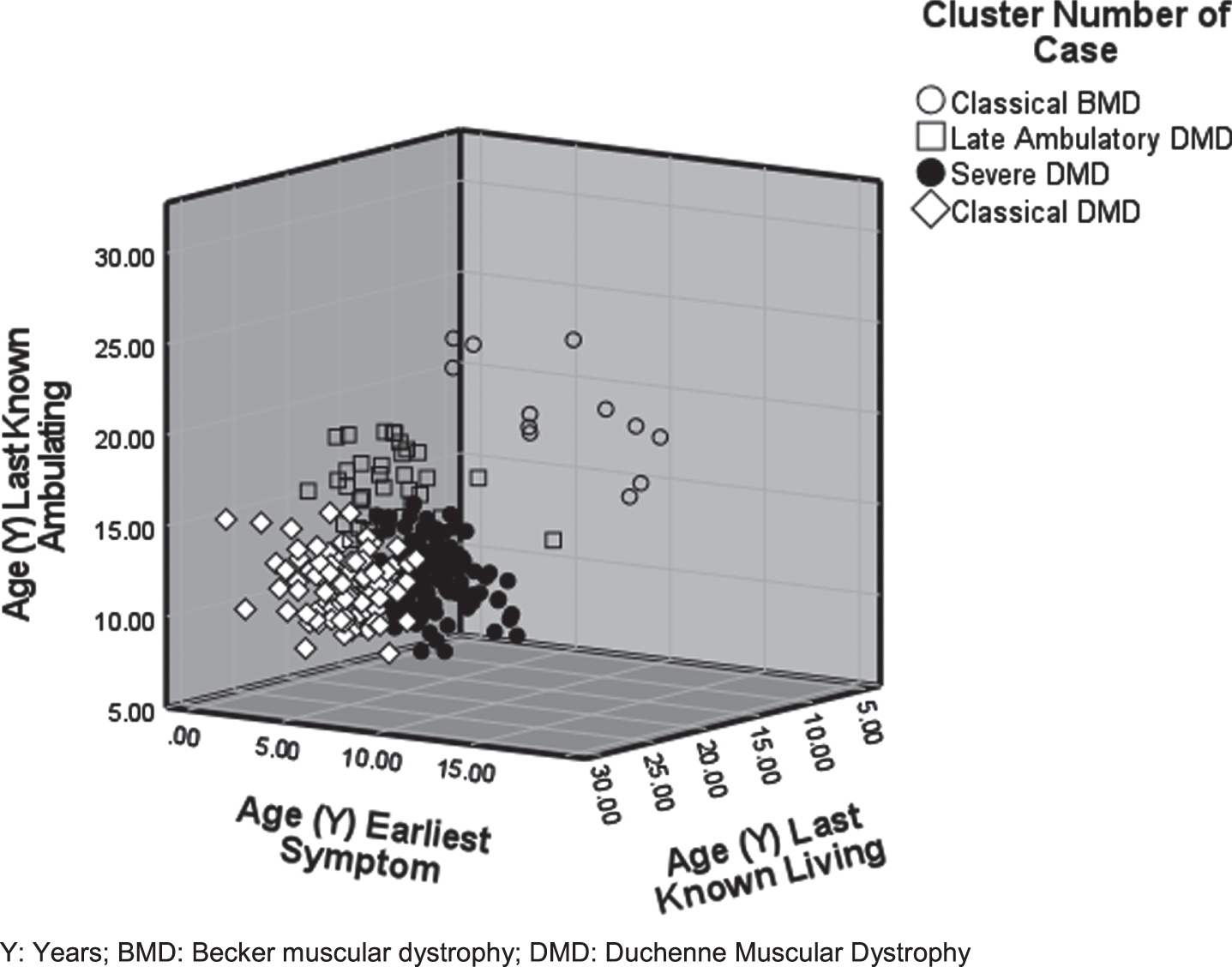
Four distinct phenotypic clusters –Classical BMD, Late Ambulatory DMD, Severe DMD, and Classical DMD –were produced through K means cluster analysis. Data include individuals who are younger than the mean event age of the additional clinical characteristics measured and therefore may represent a bias towards milder severity.
Table 1 presents the mean ages of events for each of the four clusters for the index variables used to create the clusters and the clinical variables used to characterize the clusters. Figure 1 demonstrates a 3D visualization of the clusters across these variables. The first cluster, which we named ‘Classical BMD’ (n = 12), was characterized by later ages of last known ambulation (range = 16.51–27.03 years) with 91.7% still ambulating by the end of the data collection period, later symptom onset (range = 10.17–19.18 years), and 100% living with a wide range of age at last visit (range = 16.52–27.03 years). All 12 individuals were classified as BMD according to the MD STARnet phenotype classification (data not shown).
Number of MD STARnet cases and clinical variables for the analysis and by K-means clustering
§No missing data for classification variables; MD STARnet: Muscular Dystrophy Surveillance Tracking and Research Network; DMD: Duchenne muscular dystrophy; BMD: Becker muscular dystrophy; CM: Cardiomyopathy; NIV: Noninvasive ventilation. NC: not calculated.
The remaining clusters further characterized the DMD phenotype. The ‘Classical DMD’ cluster (n = 92; 36.3% of DMD phenotype) had an average age of last known ambulation less than 11 years of age (range = 5.50–14.15 years) with 1.1% still ambulating at the date of last clinic visit and early symptom onset (range = 0.06–6.91 years). In this cluster, 66.3% were still living with a wide range of age at last visit (range = 17.36–29.45 years). The average age at the last known visit date for those not deceased was 21.61 years. Compared to the MD STARnet phenotype classification [9], 96.7% of cluster members were classified as having a DMD phenotype and 0.3% were not classifiable suggesting minimal variability within this cluster. The ‘Late ambulatory DMD’ cluster (n = 34; 13.4% of DMD phenotype) was defined by a later average age of last known ambulation (range = 12.58–19.17 years) with 29.4% still ambulating at the date of last visit and early symptom onset (range = 0.15–10.00 years). This cluster had a higher survival rate (82.3% still living) than the ‘Classical DMD’ cluster with a wide range of age at last visit (range = 12.88–24.34 years). The average age at the last known visit date for those not deceased was 17.62 years. Substantial clinical variability within this cluster is suggested by the higher percentage of individuals who were not classifiable using the MD STARnet phenotype classification [9] (BMD [n = 1], DMD [n = 17], and not classifiable [n = 16]). The last cluster, ‘Severe DMD’ (n = 127; 50.2% of DMD phenotype), was defined by an average age of last known ambulation less than 11 years (range = 5.50–14.15 years) with 9.4% still ambulating at the date of last visit, early symptom onset (range = 0.11–4.81 years), and 89.8% still living with a wide range of age at last visit (range = 12.88–24.34 years). The average age at the last known visit date for those not deceased was 13.28 years. Minimal variability within this cluster was suggested by the MD STARnet classification phenotype [9] (99.2% DMD; 0.8% not classifiable).
Cluster comparisons on clinical variables
Clinical indicators of disease severity (CM and NIV) were generally less common and occurred later within the ‘Classical BMD’ or ‘Late Ambulatory DMD” clusters than the ‘Classical DMD’ cluster (Table 1). The ‘Severe DMD’ cluster showed lower frequencies, but earlier onset, of CM and NIV relative to the other clusters. Cox proportional hazards modeling showed higher annual hazards of developing CM among those in the ‘Severe DMD’ cluster compared to the ‘Classical BMD’, ‘Late Ambulatory DMD’, and the ‘Classical DMD’ clusters (Fig. 2). CM annual hazards ranged from 1.77 (95% confidence interval [CI] = 1.07, 2.93) to 3.80 (95% = 1.41, 10.23) times higher among those with ‘Severe DMD’. Among all pairwise comparisons, the ‘Late Ambulatory DMD’ and ‘Classical DMD’ clusters showed elevated hazards of CM compared to the ‘Classical BMD’ cluster, but the confidence intervals contained the null. Similar results were found for the CM comparison between the ‘Classical DMD’ and ‘Late Ambulatory DMD’ clusters. For NIV, the parameter estimates for the ‘Classical BMD’ cluster were unreliable due to zero counts in this cluster. For the remaining clusters, annual hazards for NIV were higher among those assigned to the ‘Severe DMD’ cluster compared to the ‘Classical DMD’ (HR = 9.04, 95% CI = 3.79, 21.56) and ‘Late Ambulatory DMD’ (HR = 6.35, 95% CI = 3.51, 11.49) clusters. Lower annual hazard for NIV was found for the ‘Late Ambulatory DMD’ cluster compared to the ‘Classical DMD’ cluster, but the confidence interval contained the null.
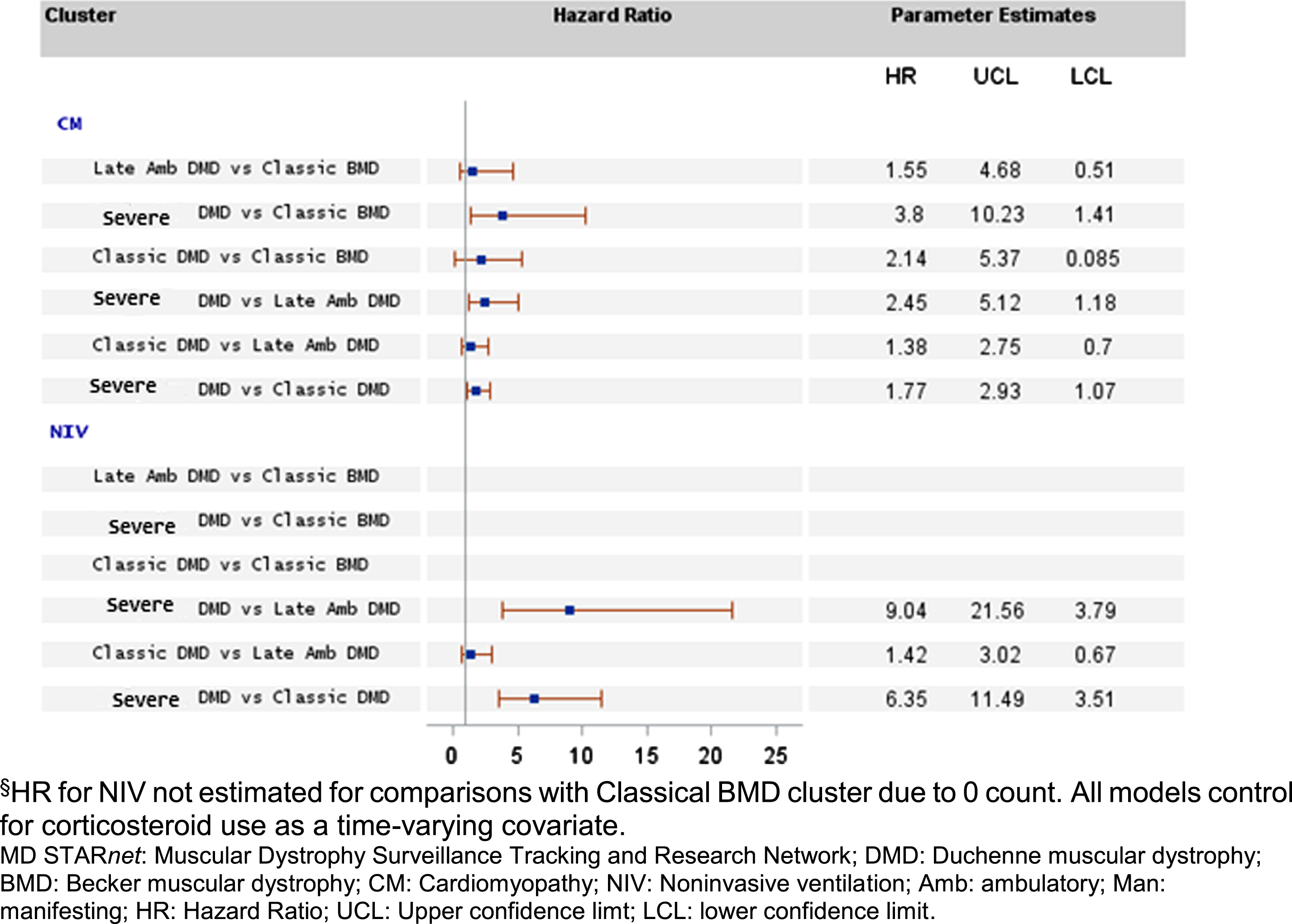
Proportional hazard ratio (HR) pairwise point estimates and 95% confidence interval demonstrating risk of earlier onset for CM and NIV by MD STARnet cluster.§
We analyzed the distribution of specific mutations across clusters. Mutations occurring in more than one individual in each cluster occurred in exons 2–20 and 44–52, in keeping with known deletion hotspots. However, there were significant differences in the distribution of certain deletions and duplications among the clusters. Deletions in exons 3–7 were only present in the ‘Late Ambulatory DMD’, deletions in exons 45–47 were only present in ‘Classical BMD’, and duplications in exon 2 were only present in the ‘Severe DMD’ cluster (data not shown). We compared the distribution of exon skippable groups by exon 44 and exon 53, specifically, and by any exon skippable mutation (cureduchenne.org) among clusters and found no significant differences (data not shown). The frequencies and percentages of frameshift mutations and mutation types across clusters are available in Supplementary Table 1.
DISCUSSION
In this study, we identified four clusters consistent with two classical phenotypes (DMD and BMD) and two potential DMD subphenotypes (‘Severe’ and ‘Late Ambulatory’) using a full spectrum of dystrophinopathy clinical symptoms. We were also able to describe cluster differences in clinical course by describing survival rates comparing onset of cardiorespiratory complications. Our results from the cluster analysis provide a classification scheme that balances the need for dichotomous phenotypic distinctions, while retaining potential subphenotypes that would allow for more refined classifications for use in research studies or clinical prediction.
Our analysis produced two clusters that corresponded with ‘Classical BMD’ and ‘Classical DMD’ phenotypes reported in the literature and considered to be the standard clinical phenotypes. Our ‘Classical BMD’ cluster demonstrated first symptoms presenting after age 10 years, ambulation ceasing after age 16 years, and no increased risk for premature death in the second decade of life. Deletions of exons 45–47 were only present in our ‘Classical BMD cluster’. This mutation has been reported to house a pseudoexon that triggers a more severe phenotype that was not present in our sample [12]. Our ‘Classical DMD’ cluster demonstrated symptom onset prior to age 5 years, early loss of ambulation, and increased risk for premature death in keeping with the current estimates of age at death [13].
The remaining two clusters corresponded to potential DMD subphenotypes. Although our ‘Late Ambulatory DMD’ cluster is consistent with intermediate MD, severe BMD, or mild DMD as described in the literature [2, 14–26], we considered this cluster to represent a subphenotype of DMD due to ages similar to those observed for the ‘Classical DMD’ cluster for first motor symptom, CM, and death. The main clinical variation that distinguished between the two former clusters was the documented age at loss of ambulation, which ranged from 12 to 16 years for the ‘Late Ambulatory DMD’ cluster and was 5.17 years later than the comparable age in our ‘Classical DMD’ cluster. However, the percentage of corticosteroid users was also higher in the ‘Late Ambulatory DMD’ cluster than that found for the ‘Classical DMD’ cluster, which could explain, in part, the defining characteristic of prolonged ambulation in the former cluster. Deletion of exons 3–7 was found only in the ‘Late Ambulatory DMD’ cluster which is in keeping with previous findings [27]. Clinically, these individuals need to be monitored carefully for complications related to long-term steroid use. These individuals could also skew clinical trials research by demonstrating positive outcomes related to the preservation of ambulation that may not be attributable to the intervention.
The ‘Severe DMD’ cluster identified in our analysis did not differ meaningfully from the ‘Classical DMD’ cluster for age at first symptom or age at loss of ambulation but did show earlier manifestations for age at death and both clinical variables (CM, NIV) despite the earlier age at last clinic visit. Humbertclaude et al. (2012) likewise proposed the presence of a severe phenotype when assessing subgroups classified according to loss of ambulation age cutoffs using interquartile ranges. They found significant correlations between the age at loss of a variety of motor function abilities and the age at which loss of ambulation occurred, as well as significant differences in pulmonary function over time. Another study identified a subset of severely affected individuals with DMD who died in their teens, none of whom were using NIV [28]. Similarly, Desguerre et al (2009) reported an ‘early infantile cluster’ with earlier symptom onset (< 2 years), an increased proportion of cases with cardiomyopathy prior to the age of 10, a significant increase in intellectual disability compared to children with Classical DMD and had significantly more risk for needing NIV than all other groups. [3] Duplication of exon 2 was unique to the ‘Severe DMD’ cluster in our sample and is reported to always predict DMD in the literature [29].
A limitation of our data is reliance on clinical measurements extracted retrospectively from medical records. While much of dystrophinopathy care is provided by centralized, multidisciplinary teams, it is possible that components of care, e.g., cardiac follow-up, may be fragmented and all records may not be available for review. Further, the timeframe over which longitudinal surveillance is conducted may limit record access for the oldest individuals in our cohort. Similarly, transitions from paper medical records to electronic medical record systems may further restrict access to earlier records. The strengths of our analysis include data derived from a large population-based cohort and a surveillance methodology that collected data longitudinally, which permitted evaluation of disease progression across the lifespan. Further, clinical and diagnostic data were individually reviewed by trained clinicians to verify a diagnosis of pediatric-onset dystrophinopathy. Finally, we were able to compare our clusters with phenotypes assigned from molecular and clinical indicators using clinical definitions representative of current research.
CONCLUSIONS
Overall, our cluster analysis provides support for variability across and within phenotypes with important implications for clinical care and research studies, especially clinical trials that rely on homogenous patient groups. In addition to the classic DMD and BMD phenotypes, we provide further evidence for ‘Severe DMD’ and ‘Late Ambulatory DMD’ subphenotypes described previously by other researchers [3, 28]. We found DMD mutations unique to three of the subphenotypes that should be explored further. These findings support the benefit of identifying DMD subphenotypes when evaluating clinical trial outcomes or findings from observational studies and providing anticipatory guidance to affected individuals and families. However, the ability to tailor a clinical approach to each dystrophinopathy subphenotype will remain limited until more consistent definitions of the phenotypic spectrum emerge along with more accurate predictions of the disease course in affected individuals. Identifying systematic, valid, and reliable means for characterizing dystrophinopathies and predicting their clinical course is a research area that needs further attention.
Footnotes
ACKNOWLEDGMENTS INCLUDING SOURCES OF SUPPORT
This work was supported by Centers for Disease Control and Prevention [DD000830, DD000831, DD000832, DD000834, DD000835, DD000836, DD000837]. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention. Research was performed in compliance with guidelines on human subjects research either through local Institutional Review Board approvals or exemptions as public health related activities. This analysis has been replicated by Molly Lamb and Kristen Conway.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.