
Abstract
While skeletal muscle remodeling happens throughout life, diseases that result in its dysfunction are accountable for many deaths. Indeed, skeletal muscle is exceptionally capable to respond to stimuli modifying its homeostasis, such as in atrophy, hypertrophy, regeneration and repair. In particular conditions such as genetic diseases (muscular dystrophies), skeletal muscle’s capacity to remodel is strongly affected and undergoes continuous cycles of chronic damage. This induces scarring, fatty infiltration, as well as loss of contractibility and of the ability to generate force. In this context, inflammation, primarily mediated by macrophages, plays a central pathogenic role. Macrophages contribute as the primary regulators of inflammation during skeletal muscle regeneration, affecting tissue-resident cells such as myogenic cells and endothelial cells, but also fibro-adipogenic progenitors, which are the main source of the fibro fatty scar. During skeletal muscle regeneration their function is tightly orchestrated, while in dystrophies their fate is strongly disturbed, resulting in chronic inflammation. In this review, we will discuss the latest findings on the role of macrophages in skeletal muscle diseases, and how they are regulated.
ABBREVIATIONS
AMP-activated Protein Kinase
Bone Marrow
Becker Muscular Dystrophy
Bone Marrow Derived Macrophages
CCAAT-enhancer-binding proteins
C-C chemokine ligand
C-C chemokine receptor
Creatine Kinase
Colony Stimulating Factor
Connective Tissue Growth Factor
C-X-C chemokine ligand
C-X-C chemokine receptor
Damage-Associated Molecular Pattern
Dystrophin-Glycoprotein Complex
Diphtheria Toxin A
Extracellular Matrix
embryonic myosin heavy chain
Fibro/Adipogenic Progenitor
Granulocyte Receptor
Granulocyte Macrophage
Interferon Gamma
Insulin-like Growth Factor
Interleukin
Limb-girdle Muscular Dystrophy
Latent TGFβ Binding Protein
lipopolysaccharide
Mitogen-activated protein kinase
Myosin Heavy Chain
Mitogen-activated protein kinase phosphatase 1
Matrix Metalloproteinase
Muscular dystrophy
Myositis-specific antibody
Nicotinamide phosphoribosyltransferase
Nuclear Factor I X
Nuclear Factor kappa B
Nonsteroidal anti-inflammatory drugs
Osteopontin
Platelet Derived Growth Factor Receptor
receptor activator of nuclear factor NF-κB ligand
Regulated upon Activation, Normal T Cell Expressed and Presumably Secreted
Satellite Cell
sarcoglycan
Transforming growth factor beta
T Cell Immunoglobulin And Mucin Domain Containing 4
Tumor Necrosis Factor
regulatory T cell
urokinase plasminogen activator
Vascular endothelial growth factor
Wild Type
INTRODUCTION
Disruption of homeostasis in skeletal muscle tissue can be triggered by many variables, including changes in diet or exercise, local injury, systemic infection, genetic disease, and ageing. In most of these cases, inflammation and in particular macrophages play a primordial role in the pathology. Macrophages, along with neutrophils, are part of the leukocyte family first described in 1908 by Elie Metchnikoff. In addition to their immunoprotective role against pathogens, macrophages have a number of additional roles during development and tissue remodeling, in particular in skeletal muscle [1–3].
Skeletal muscle regeneration is made possible by a population of adult muscle stem cells called satellite cells (SC) [4]. After damage or microenvironment changes, SCs activate, proliferate, and differentiate into myoblasts/myocytes before fusing with one another or with surrounding myofibers. As fitting their definition as a stem cell, SCs are capable of self-renewal which maintains a potent pool of cells throughout life (reviewed in [5, 6]). Despite the essential role SCs play in muscle regeneration, these events won’t happen without the help of other cells harbored in the interstices between the myofibers: endothelial cells, pericytes [7], fibro/adipogenic progenitors (FAP) [8–10], and foremost, macrophages [11–16]. Specifically, following damage, macrophages are responsible for the release of a pool of cytokines, chemokines and alarmins called damage-associated molecular pattern (DAMPs). These molecules are part of a coordinated response that initiates sterile inflammation and induction of blood-circulating monocytes’ infiltration into the tissue.
In healthy tissue, successful skeletal muscle regeneration is typically complete within 3 weeks, and without any need for external intervention (anti-inflammatory drugs for example) [17]. However, when disrupted, the process can take months and is often associated with fibrotic deposition (scarring) and adipogenesis. Examples of such disruptions in muscle homeostasis include repeated injures, volumetric mass injuries, modification in homeostasis during ageing (sarcopenia), prolonged immobility/atrophy (e.g. cancer: cachexia), genetic diseases that directly affect the muscle (e.g. Duchenne and Becker Muscular Dystrophies (DMD, BMD), and Limb-girdle muscular dystrophy (LGMD)).
In this review, we briefly describe the roles of macrophages in skeletal muscle regeneration, then turn our focus to their roles in muscular dystrophies (MD) and how current treatments act on their functions.
MACROPHAGES, DEFINITION AND FUNCTION IN SKELETAL MUSCLE REMODELING
To study muscle regeneration and inflammation, injury models that make use of toxins (notexin, cardiotoxin), chemicals (barium chloride) and mechanical trauma (crush, freezing, ischemia, laceration) are used to stimulate a response. Depending on the type of damage the kinetics of the inflammatory response will vary, however the overall regenerative process of the tissue remains the same (for more information about injury models, please read [18]).
The study of macrophages in vitro is usually conducted using bone marrow-derived macrophages (BMDM). Macrophage polarization in vitro is required to induce the secretion of specific cytokines associated to their functions such as phagocytosis of apoptotic cells, cell growth and tissue repair promotion, or fighting bacterial infections. Like T cells, macrophages can acquire two main inflammatory profiles: classically activated M1 (related to type 1 inflammation –Th1) and alternatively activated M2 (related to Type 2 inflammation –Th2). The Th1/Th2 paradigm in macrophage has been extensively discussed and we encourage reading the following reviews [19, 20]. BMDM are stimulated with IFNγ, IL-4/IL-13, or IL-10, to respectively mimic either the pro-inflammatory/classically activated state “M1”, the alternative activation state “M2a”, or the anti-inflammatory state “M2c” [21]. Other M2 states have been proposed, such as M2b (immune complex activation [22]) and M2d (TLR antagonist [23, 24]), but are not relevant in muscle biology. Moreover, Lipopolysaccharide (LPS) alone or together with IFNγ, can be used to induce a stronger pro-inflammatory activation state. However, in the case of tissue regeneration and sterile inflammation, the use of LPS and of these in vitro methods in general might push macrophages into state not representative of that found in vivo.
The understudied tissue-resident macrophages
Skeletal muscle resident macrophages are quiescent cells, occupying space within the connective tissue that surrounds myofibers and in close proximity to blood vessels [25, 26]. As in other tissues, distinct subsets of resident macrophages arise from either developmental origins (yolk sac-, aorta-gonad-mesonephros- (AGM), or liver-derived hematopoiesis) or from the adult bone marrow (BM) [27]. It has been shown that tissue-resident macrophages acquire tissue specific functions. For example, Kupffer cells (liver) play a crucial role in the clearance of blood toxins, and alveolar macrophages (lung) will actively clean pathogens and microorganisms from the airways [28–30]. In skeletal muscle, so far the only proposed role of tissue resident macrophages is the attraction of circulating blood monocytes to the site of damage [26]. Yet, tissue resident macrophages are still heterogeneous, with a sub-type expressing stress-response genes such as Klf2 or Fos.
It has been established that tissue-resident macrophages derive from primitive hematopoiesis in the liver and definitive hematopoiesis in the BM [32, 33]. As of now, the muscle research community lacks a specific marker to distinguish skeletal muscle resident macrophages from infiltrating monocytes. However, it seems that a proportion of resident macrophages are embryonically derived, while another subpopulation is maintained by blood-derived monocytes [31, 34]. The embryonic-derived tissue-resident macrophages are Ly-6CCCR2F4/80hiCD11blow, while the BM-derived tissue-resident macrophages are Ly-6CCCR2F4/80lowCD11bhigh [32] (Table 1). More recently, the marker Lyve1 has been added to the list of the markers for tissue resident macrophages [31, 35]. Wang et al., also suggest that Lyve1 expression (high versus low) could differentiate between the embryonic-derived (Lyve1low) and the blood-derived (Lyve1high) tissue resident macrophages (Table 1). [31] In the heart, tissue-resident macrophages can be distinguished by expression of the marker T cell Immunoglobulin and Mucin Domain containing 4 (TIMD4). TIMD4+ macrophages are able to locally self-renew, while TIMD4 cells are replaced by blood-derived cells [36]. Moreover, cardiac resident macrophages seem to have very specific functions depending on their location. For example, resident macrophages present in the atrioventricular nodes are required to maintain cardiac contraction via the formation of gap junctions with cardiomyocytes [37]. The regulation, self-renewal, and function of skeletal muscle-resident macrophages at homeostasis and during regeneration is not yet understood. The characterization (ontology and specific markers) of this population should be definitively one of the focus points for better understanding of their function at steady state, during tissue remodeling (mild or acute damage), ageing, and in disease [2, 30].
Monocyte/Macrophage populations
Infiltrating monocytes and their function as macrophages
Circulating blood-monocyte infiltration
In blood, monocytes separate into two subsets [38]. The first, which is CCR2Ly-6CCX3CR1highCD11b+F4/80+ functions to patrol the vasculature in search of pathogens [39] (Table 1). The other subset is CCR2+Ly-6C+CX3CR1lowCD11blowF4/80low and homes to damaged tissues primarily through the CCL2(MCP1)-CCR2 axis [40–44]. This cytokine-receptor interaction was one of the first described to induce monocytes infiltration into tissues after damage and has been observed as indispensable in liver, heart, and skeletal muscle [43, 45–47]. Since then, other chemokines have been described, such as: RANTES/CCL5, MIP3/CCL3, MIP4/CCL4, MCP-3/CCL7, MCP-4/CCL8 [48]. Sources of these chemoattractant can vary by tissue and context. For example, deletion of tissue resident FAPs using the PDGFRα-CRE:DTA mouse model induced a strong reduction in infiltrating CD45+ cells after damage in skeletal muscle [49]. While this has not been backed up with migration assays in vitro, FAPs are known to produce and secrete MCP-1 and CSF1, which are known chemoattractant for leucocytes [50]. Alternatively, cells such as SCs and myofibers have also been shown to attract monocytes/macrophages [51, 52]. Currently, only CCR2+Ly-6C+CX3CR1lowCD11blowF4/80low monocytes are known to infiltrate damaged muscle [11, 54]. One of the best pieces of supportive evidence comes from the use of the Nur77-KO mouse model. Nur77 (also known as NR4A1) is an orphan transcription factor involved in cell proliferation. Depletion of Nur77 induces a block in S phase, leading to apoptosis of Ly6C MO in the bone marrow [55]. Interestingly, while Nur77-KO animals lack the Ly-6C blood-circulating monocyte population, Ly-6C macrophages are found in the muscle after acute damage in similar numbers to control littermates [53].
Blood-derived macrophage functions
The infiltration of monocytes and their differentiation into macrophages is essential for proper skeletal muscle regeneration [11, 56–58]. Once extravasated, blood monocytes differentiate into inflammatory macrophages and lose CCR2 expression. Of note, this differentiation step is not fully understood yet and may be independent of the process leading to the appearance of alternatively activated and pro-regenerative macrophages (skewing), which is associated with the downregulation of Ly-6C [11, 53] (Fig. 1). Ly-6C+ and Ly-6C– macrophages are observed in a temporally precise sequence and are efficiently coordinated for skeletal muscle regeneration.
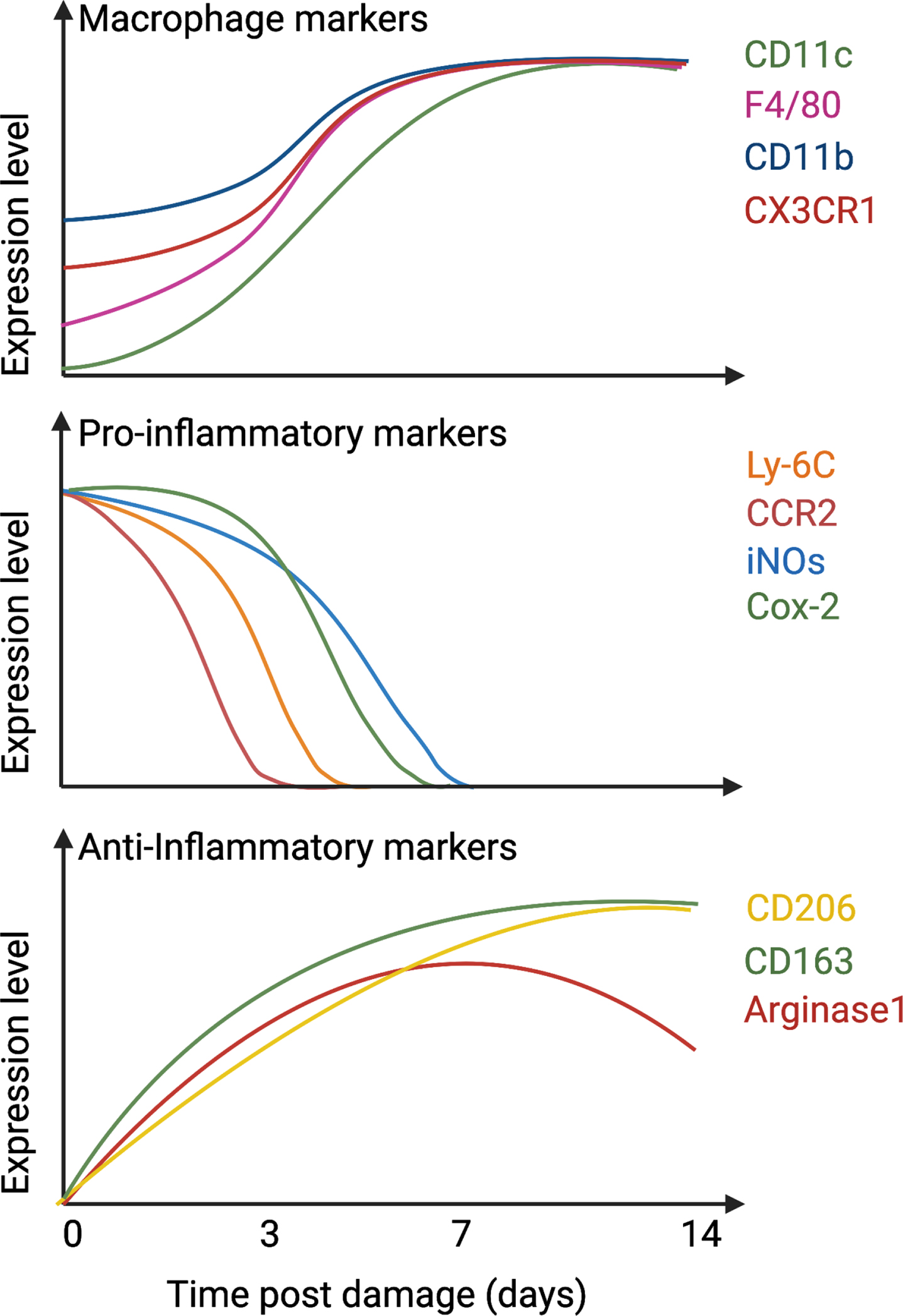
Temporal expression of macrophage and inflammatory markers. After damage, infiltrated monocytes differentiate into macrophages, up-regulate CD11c, CD11b, F4/80, and CX3CR1 (top graph) and express pro-inflammatory markers such as Ly-6C, CCR2, iNOs, and Cox-2 (middle graph). After 1-2 days in the tissue, they downregulate pro-inflammatory markers and start to express anti-inflammatory proteins such as CD206, CD163, and Arginase 1 (bottom graph).
Pro-inflammatory macrophages (Ly-6C+F4/80+CD11b+CX3CR1low) secrete cytokines and growth factors that support SC proliferation but induce death in FAPs [52, 59]. Following the clearance of debris, dead cells, and necrotic myofibers, macrophages slowly activate a program that skews them towards a pro-regenerative phenotype (Ly-6C–F4/80+CD11b+CX3CR1high), which supports myogenic cell differentiation and fusion [17, 60–62], and FAP survival [56, 59]. Knockout murine models have demonstrated the importance of this phenotypical skewing for efficient muscle regeneration and identified several actors guiding this process, including AMP-activated Kinase (AMPK), Mitogen-activated protein kinase (MAPK) phosphatase 1 (MKP-1), CCAAT-enhancer-binding proteins (C/EBPβ), and Nuclear Factor I X (Nfix) [17, 63–65]. To note, in addition to Ly-6C, F4/80 and CD11b, other markers can also be used to distinguish pro-inflammatory macrophages from the pro-regenerative type and are highlighted in Fig. 1 and Table 2. Through this process, macrophages are sensitive to and secrete various cytokines required for efficient skeletal muscle regeneration, briefly described here (Fig. 2):
Markers for ma macrophages involved in skeletal muscle regeneration
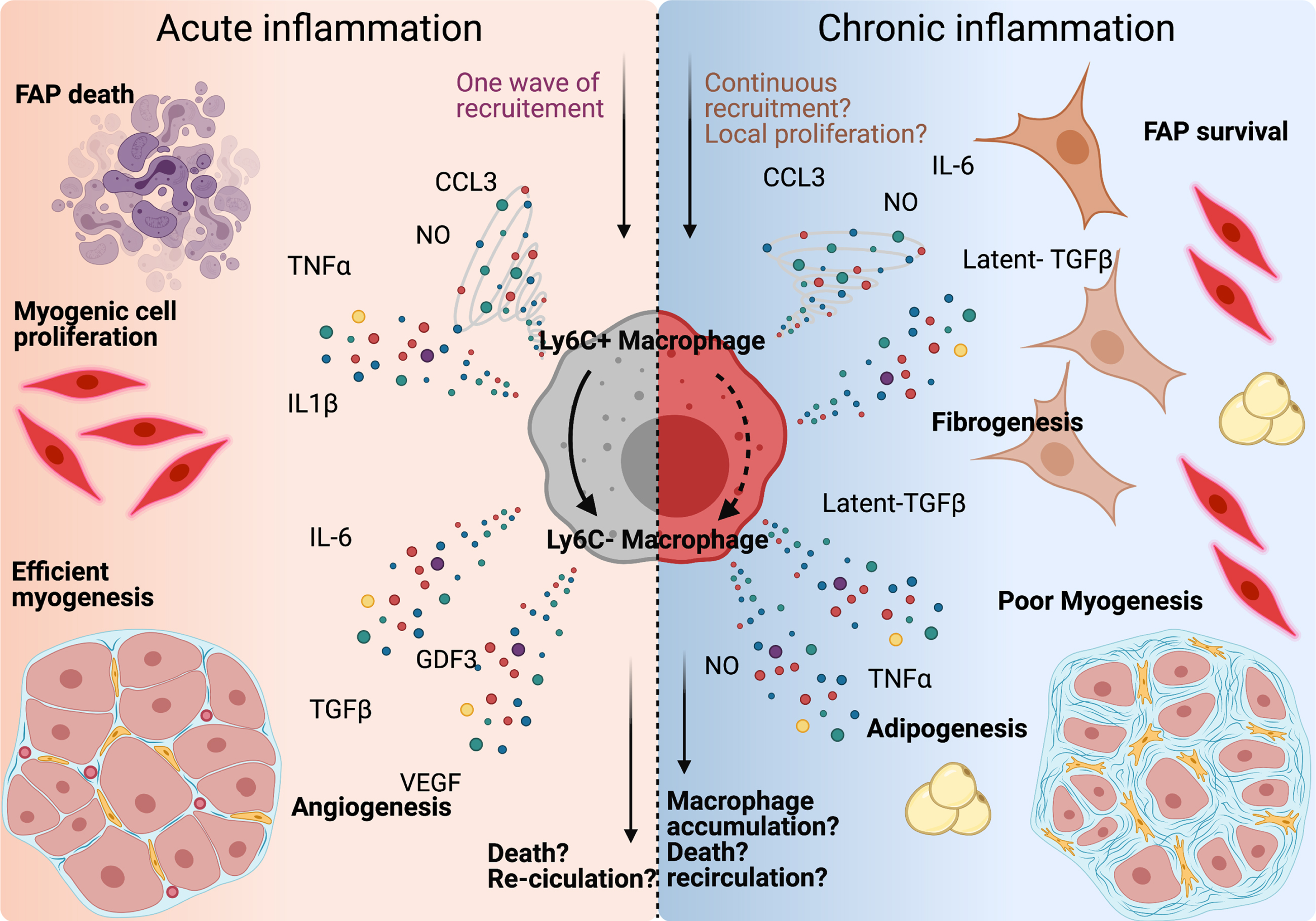
Macrophages orchestrate muscle-resident cell behaviour during tissue repair. Left panel: after injury, Ly-6C+ macrophages secrete CCL3, NO, TNFα and IL-1β, clear fibro-adipogenic progenitors (FAPs) from the tissue, and support myogenic cell proliferation. Once skewed to an anti-inflammatory profile, macrophage produce IL-6, GDF3, TGFβ, and VEGF, which participate into the support of the myogenic program and myofiber growth. Macrophage content within the tissue returns to basal around 2 weeks after damage by either re-circulation or local apoptosis. Right panel: in case of repeated trauma, the number of macrophages present in the tissue rises, which could be due to continuous infiltration, or to local proliferation. Both Ly-6C+ and Ly-6C–macrophages are present within the tissue, which causes the accumulation of both pro- and anti-inflammatory cytokines in the damaged area. FAPs are activated, differentiate into fibroblast and adipocytes and myogenesis is delayed.
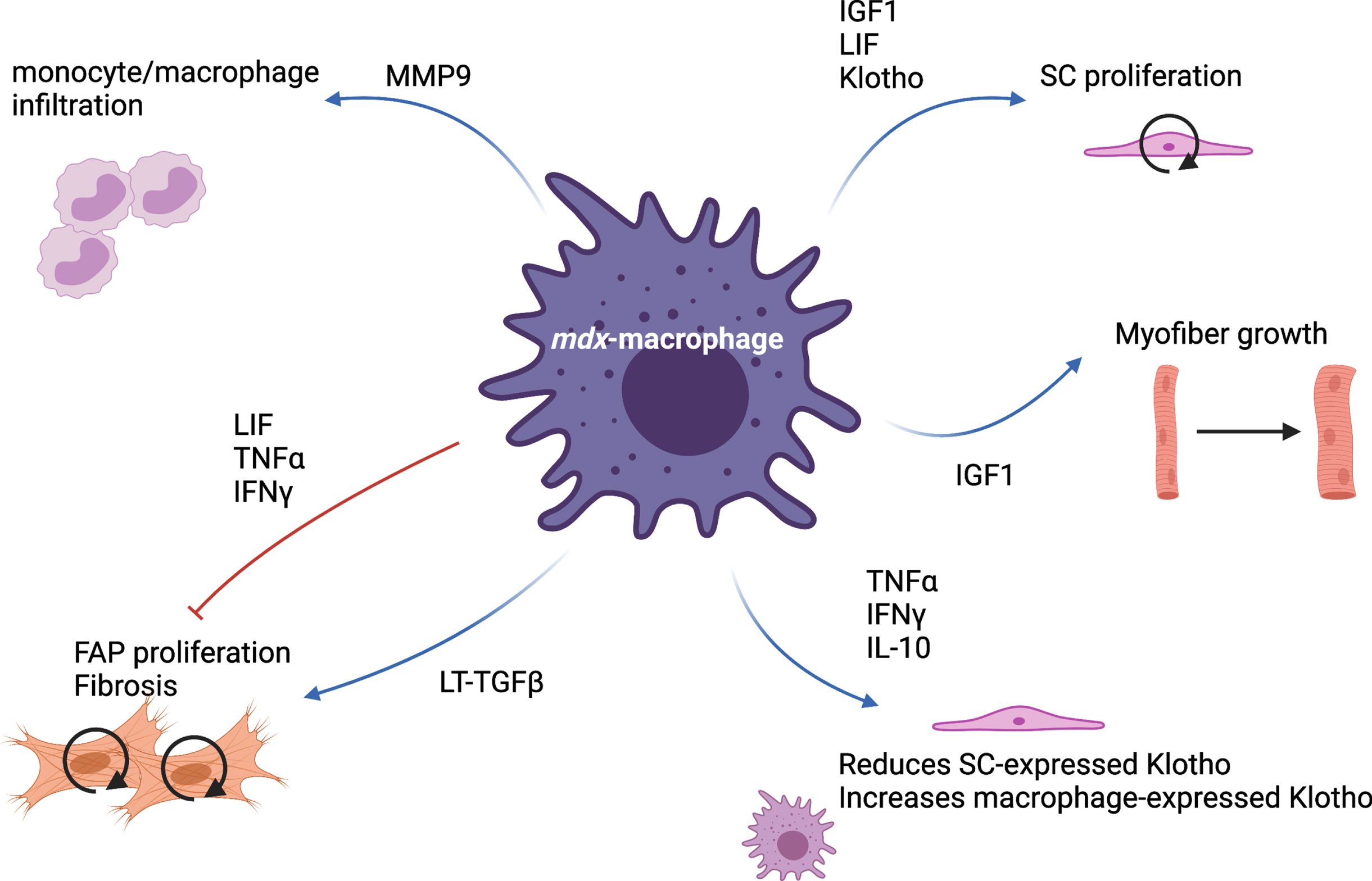
mdx-macrophage specific functions. Via the production of many cytokines and proteins, macrophages present in mdx muscle are able to simultaneously stimulate and inhibit various cellular processes such as monocyte infiltration, satellite cell (SC) proliferation, myofiber growth, fibro-adipogenic progenitor (FAP) proliferation and differentiation.
HISTOPATHOLOGY AND ANIMAL MODELS OF MUSCULAR DYSTROPHIES
MDs are a heterogeneous group of inherited disorders characterized by progressive wasting and weakness of muscle tissue that compromises patient mobility, leading to wheelchair dependency. In severe cases, patients with MDs die prematurely due to respiratory and cardiac failure [99]. Many MDs are caused by mutations in the genes coding for proteins of the dystrophin-glycoprotein complex (DGC) or required for the correct assembly of the DGC. The main components of this multiprotein complex are dystrophin and sarcoglycan subunits. Structurally, the DGC links the F-actin cytoskeleton of myofibers to the ECM. The absence of even one protein of the DGC often causes the disassembly of the entire complex, causing sarcolemmal (the myofiber plasma membrane) fragility and leading to myofiber damage and necrosis that is aggravated by contractile activity [100–102]. Damaged myofibers are repaired or replaced by SCs, but as they share the same genetic mutation, the newly formed myofibers are destined for the same degenerative fate. Consequently, muscle tissue enters into a continuous cycle of degeneration and regeneration that results in chronic inflammation and substitution of contractile muscle tissue with adipose and fibrotic tissue. At the histological level, dystrophic muscle is characterized by necrotic myofibers, desynchronized centrally-nucleated regenerative myofibers, immune cell infiltration, and the presence of fatty/fibrotic lesions in place of muscle tissue [103].
The most common MD is the Duchenne muscular dystrophy (DMD), an X-linked autosomal recessive disease caused by a mutation in the ∼2.4 Mb dystrophin gene that results in complete loss of the dystrophin protein and affects approximately 1 in 3,500–6,000 boys [104, 105]. The first signs of disease are usually observed around 2 to 3 years of age, followed by progressive muscle wasting, gradually leading to wheelchair use and eventual death caused by respiratory and cardiac complications [99, 107]. In DMD 60%of dystrophin mutations are large insertions or deletions that lead to frameshift errors downstream, whereas approximately 40%are point mutations or small frameshift rearrangements [108]. Becker-type muscular dystrophy (BMD) is also caused by a mutation to the dystrophin gene, but one that permits the synthesis of an internally truncated and partially functional protein, leading to a milder phenotype in affected boys [99]. MDs are particularly difficult to treat due to the post-mitotic nature of cardiac and skeletal muscle, as well as the abundancy of muscle tissue across the body. Different animal models are used to understand the development of the dystrophic disease as well as molecular and cellular pathways involved in this process [109]. Here are some of the main mouse models:
In mdx, sgca(-/-), and DMD, a highly inflammatory environment (in terms of cell infiltration and cytokine detection) is observed compared to WT, non-damaged muscles [111, 134–137]. While macrophages are required and beneficial for efficient muscle regeneration after acute injury (see chapter above), chronic inflammation leads to tissue damage. In the case of skeletal muscle, the deleterious effect of macrophages during periods of chronic damage is hypothesized to be attributed to: 1) a sustained inflammatory environment that promotes muscle tissue damage, and 2) fibrosis induced by abnormal persistence of wound-healing macrophages. Indeed, in the last decades, many studies thoroughly investigated the phenotype and functions of macrophages in MDs.
ROLE OF MACROPHAGES IN CHRONIC INJURY: FRIENDS OR FOES?
Muscle regeneration following acute injury can be impaired by genetically or pharmacologically affecting macrophage’s capacity to infiltrate the tissue [11, 138–140]. However, in dystrophic muscle the decreased inflammation associated with modifying macrophage’s infiltrative capacity often correlates with improved tissue functionality. In both human DMD, and mouse models like in sgca(-/-) and mdx, the number of macrophages correlate with expression of fibrotic markers [56].
Depletion of blood-circulating monocytes in young mdx mice using an anti-F4/80 antibody reduces the number of damaged myofibers, presumably because it delays the peak of inflammation that normally occurs in mdx mice at early time point [141] (Table 3). Preventing macrophage infiltration by deleting the TLR4 receptor also attenuates MD progression in 6 and 12-week-old mdx mice by limiting muscle damage and fibrosis, while also limiting loss of force [142] (Table 3). Likewise, deleting the CCR2 receptor in mdx mice (mdx:CCR2-/-) decreases macrophage infiltration in the diaphragm at 6 weeks of life. Interestingly, at 12 weeks of age, mdx:CCR2-/- mice have the same number of intramuscular macrophages compared to mdx control mice, suggesting a role for tissue-resident macrophages in disease progression. Furthermore, there are more CD206+ (pro-regenerative) macrophages and fewer iNOS+ (pro-inflammatory) macrophages in the diaphragm of mdx:CCR2-/- mice at 6 weeks of age. Again, this may be due to contribution of tissue-resident macrophages, which may not be able to efficiently acquire a pro-inflammatory phenotype. No difference in terms of histopathology is observed between mdx and mdx:CCR2-/- mice at 6 weeks of age, but at 12 weeks of age a decrease in the number of necrotic myofibers and area of fibrosis is observed in the diaphragm of double mutant mice. Functional improvement in the diaphragm is observed at both 6 weeks and 12 weeks of age [143] (Table 3).
Depletion of monocyte/macrophage in mdx mouse model
Consistent with these results, the use of a pharmacological antagonist of CCR2/CCR5 receptor (CVC or cenicriviroc) in mdx mice from 2- to 6-weeks of age decreased macrophage infiltration of the diaphragm in treated mice without any effect on myofiber necrosis and fibrosis. However, no changes in the ratio of CD206+ or iNOS+ macrophage were observed [144] (Table 3). Analysis of mdx mouse pathology at 1 year shows that these animals approach a similar disease state to that of human DMD patients (force, regenerative capacity, central nuclei, muscle hypertrophy, and myofiber branching). Similarly, the early improvements observed in the diaphragm and quadriceps of mdx:CCR2-/- is lost by 6 months of age in these animals [145]. As in the study by Mojumbar et al., the diminution of macrophage infiltration at an early time point (4 weeks of age) disappears by 14 weeks and 6 months [143, 145], It remains unclear whether the regression in histological and functional improvement is due to re-infiltration of macrophages or from an effect on their inflammatory profile and function.
When mdx mice are crossed with a urokinase plasminogen activator (uPA) deficiency model the progeny exhibit an increase in the degenerative muscle phenotype, including increased fibrosis and decreased muscle function. This phenotype also correlates with a decrease in macrophage infiltration in the dystrophic muscle [146] (Table 3). Interestingly, the transplantation of WT-BM into the mdx:uPA-/- mice increased macrophage infiltration in dystrophic muscle and reversed the deleterious effect of uPA knockout, suggesting a pro-regenerative role of infiltrating uPA-expressing macrophages within the dystrophic muscle. Of note, muscular regeneration after acute injury is also observed in uPA-/- mice and is rescued by WT BM transplantation as well [146]. Another study demonstrated that the depletion of macrophages in mdx mice (using a mouse model expressing the Dipheteria Toxin Receptor under the CD11b promotor) compromises muscle regeneration at 12 weeks of age by promoting adipogenic fate in SCs [147].
In conclusion, the functional phenotype of macrophages and more specifically, their trophic function toward other cells within the muscle, impacts disease progression more than the overall number of macrophages present in dystrophic muscle, and this parameter should be taken in consideration when analyzing muscle histopathology (Table 3).
Trophic functions of macrophages toward muscle homeostasis in mdx mice
During healthy muscle regeneration, two functionally distinct populations of macrophages are observed in a temporally precise sequence. Together, these pro-inflammatory Ly-6C+ and pro-regenerative Ly-6C– macrophages efficiently coordinate to help heal the damage and return the affected muscle tissue back to homeostasis. However, the situation becomes more complex in dystrophic muscles, where gauging the functional status of the macrophages within the affected tissue using simple markers such as Ly-6C may be less reliable (Fig. 1 and Tables 1 2). In fact, a recent work suggests that this method fails to accurately capture the nuances of macrophage transcriptional status within these situations [56]. During muscle regeneration, macrophages simultaneously express pro- and anti-inflammatory programs, and their function is ultimately determined by the balance of these programs [17, 56] (Fig. 1 and Table 1). Gene expression analyses of Ly-6C+ and Ly-6C– macrophages sorted from non-fibrotic and fibrotic dystrophic muscle (mdx and sgca(-/-) mice) show that the canonically pro-regenerative Ly-6C– population actually express high levels of pro-inflammatory markers, suggesting the presence of a mixed-function population of macrophages within dystrophic muscle [56]. Indeed, nearly 50%of macrophages present in mdx mice express both TNFα and TGFβ [59]. Future studies that incorporate single cell and spatial RNA-sequencing technologies allow us to better understand macrophage polarization and function in dystrophic muscles and particularly, how they interact with other cell types. Nevertheless, numerous studies have helped to identify general functions of macrophages in chronic muscle injury.
Genetic ablation of IFNγ in mdx mice (mdx:IFNγ-KO) does not affect macrophage infiltration in 4 and 12 month-old mice, but does lead to a decrease in iNOS production and reduced damaged myofibers in hindlimb muscles [148]. The loss of IFNγ also increased Myod1 and Myogenin expression, which are markers of proliferating and differentiating myoblasts whose expression is linked with regeneration. Indeed, mdx:IFNγ-KO mice have more centrally-nucleated myofibers and exhibit improved muscle function, suggesting enhanced tissue repair [148].
Deletion of iNOS in mdx mice (mdx:iNOS-KO) has no effect on macrophage infiltration, but decreases myofiber lysis [149]. In DMD and mdx muscles, a local increase in TNFα has been observed, and leads to the activation of NFκB signaling [150, 151]. Consistently, NFκB activity increases in diaphragm, gastrocnemius and tibialis anterior of 5-week-old mdx mice compared to WT mice. In mdx mice, NFκB is present in nuclei of both regenerating myofibers and immune cells.
Macrophage-specific deletion of NFκB in mdx mice decreases the number of necrotic myofibers and results in reduced expression of TNFα and IL-1β in 4-week-old animals. Interestingly, the specific deletion of NFκB in myofibers stimulates muscle regeneration and leads to an increase in embryonic myosin heavy chain positive (eMHC+) myofibers. Furthermore, an increase in cells expressing Pax7 and MyoD is observed in 4-week-old mdx-NFκB-KO mice, suggesting a negative role of NFκB on SC behaviour. Finally, pharmacological inhibition of IKK/NFκB pathway reduces muscle necrosis and improves muscle regeneration, supporting the notion that this pathway is active in several cell types present in dystrophic muscle which act synergistically to rescue myopathic progression [151].
Weekly intra-peritoneal injections of TNFα-blocking antibodies in mdx mice during the first 90 days of life decrease the number of necrotic myofibers and have a positive effect on treadmill running time. Unfortunately, neither the inflammatory profile, nor possible mechanisms of action were investigated in this study [152]. The use of anti-IL-6 antibody on mdx:utrn-/- from 2 to 13 weeks of age, was shown to significantly improve skeletal muscle histopathology by reducing Creatine Kinase (CK) levels, fibrosis deposition, increasing regenerating myofiber size [153]. However, these effects were not seen in the diaphragm. Paradoxically, while IL-6 has been shown to participate to the DMD pathology [154], the inflammatory response, quantified by q-RT-PCR was unchanged [153]. Moreover, Kostek et al. treated mdx mice with IL-6 blocking antibodies for 5 weeks with no functional or histological improvements, but rather an increase in their “inflammation score” (quantified by the number of mononuclear cells observed on muscle slides) [155].
In a related recent study, 4-month-old mdx:utrn+/– mice were injected intra-peritoneally every 3 days, for 28 days, with blocking antibody against RANKL (receptor activator of nuclear factor NFκB ligand) [156]. While muscle damage and fibrosis were decreased, and associated with an increase in myofiber size, the number of infiltrated macrophages did not change after treatment. Nevertheless, a relative increase of CD206+ macrophages was observed in muscle of mdx/utrn+/– mice injected with RANKL blocking antibody [156]. Similarly, the frequency of CD206+ cells increased in muscles of mdx:IFNγ-KO mice [148].
Mdx:IL-10-KO mice have elevated numbers of necrotic myofibers and perform poorly in treadmill performance tests at both 4 and 12 weeks of age, when compared to mdx mice. These mice have a lower frequency of CD163+ pro-regenerative macrophages in muscle, but quantification of total macrophage infiltration was not performed [81]. Thus, IL-10 secretion seems to be beneficial for dystrophic muscle.
Finally, the beneficial effect of depleting specific pro-inflammatory cytokines or signaling pathways in MD pathology could be due to the fact that dystrophic myofibers are more sensitive to oxidative stress [157, 158].
Overall, the above studies suggest that decreasing macrophage pro-inflammatory signals and pushing them to an anti-inflammatory phenotype could be beneficial for dystrophic muscle [156]. However, most studies have focused on the first few weeks of life in mdx mice, which unlike human patients is known to peak in inflammation at 4-weeks of age [141, 159]. Thus, further detailed studies and critical evaluation of these datasets will be required to assess the potential of macrophage modulation as a therapeutic option for MD patients.
The role of macrophages in the formation of fibrosis in muscular dystrophies
At steady state, muscle ECM is a three-dimensional network that represents around 5%of tissue volume. The ECM is primarily composed of collagen type I, with myofibers being surrounded by collagen IV [160]. ECM’s function as a structural substrate capable of supporting muscle fibers, blood vessels, and nerves must not eclipse its critical role as a regulator of cell fate. For example, it has been demonstrated that Collagen V and VI are intimately linked to SC quiescence [161, 162]. After muscle injury, ECM is degraded by inflammatory cells, which permits their infiltration into the damaged tissue, while concomitantly facilitating effective migration of SCs [163, 164]. Specifically, the ECM is degraded by matrix metalloproteinases (MMPs), secreted by inflammatory cells and damaged myofibers [165–167]. A transient partially degraded ECM participates in myoblast differentiation and provides a scaffold for regenerative myofibers [168]. As such, the balance between ECM degradation and production is important for efficient regeneration. Fibrosis, which is a characteristic feature of MD pathology, is caused by excessive accumulation of ECM components resulting from ECM over-production, a defect in its degradation, or both [168, 169]. Among the factors that promote ECM remodeling, TGFβ is believed to be the most important. TGFβ not only induces collagen I, connective tissue growth factor (CTGF/CCN2) and fibronectin expression by FAPs/fibroblasts, but also inhibits MMP expression in fibroblasts through SMAD pathway activation [90, 170–172]. While macrophages and SCs do express some collagen proteins, FAPs are the primary source of ECM component secretion [162, 174]. FAPs were identified in 2010 and are defined as SCA-1/PDGFRα+, CD31/CD45/α7int– cells. FAPs are located in the skeletal muscle interstitial space and proliferate upon injury [8, 9]. In both in vitro and in vivo conditions, FAPs are capable of spontaneously differentiating into fibroblasts and adipocytes, but they do not differentiate into myogenic cells [8, 50]. They stimulate myogenic differentiation in SCs, which once differentiated into myofibers, block FAP adipogenic differentiation [8, 176]. In mdx mice, the number of PDGFRα+ cells positively correlates with fibrosis and addition of TGFβ1 to FAPs in vitro induces a dose-dependently increase in fibrotic markers (such as collagen I and CTGF [8, 59]), supporting the importance of this factor in fibrosis development. Importantly, the balance between proliferation and apoptosis of FAPs is directly determined by macrophage-derived TGFβ1
The unforgettable TGFβ
During skeletal muscle regeneration, pro-inflammatory macrophages first secrete TNFα, which induces FAP apoptosis bringing their numbers back to pre-damage levels. Next, pro-regenerative macrophages secrete TGFβ to stimulate the survival of remaining FAPs and the production of a regenerative provisional matrix. Proper balance and coordinated expression of these cytokines is thus critical for reestablishment of the ECM [56, 177] (Fig. 2). For more background on the role of FAPs in muscle homeostasis, we suggest the following reviews [178, 179].
In DMD patients, TGFβ1 is elevated in both blood plasma and muscle, and is correlated with fibrosis [91, 181]. In addition, treatment of WT mice with recombinant TGFβ1 stimulates the expression of collagen I and induces muscle fibrosis independently of injury or disease [182]. It has been demonstrated that asynchronous myofiber damage and regeneration, such as that observed in DMD, directly induces fibrosis through the TGFβ1 pathway [117]. However, the induction of a fibrogenic program in FAPs is not the only way in which TGFβ1 can modulate muscle homeostasis. The binding of TGFβ1 and/or myostatin to their specific cell-surface receptors (TGFBR1/ALK5 and TGFBR2 dimer for TGFβ1; activin receptor types IIA and IIB dimer, or TGFBR1/ALK5 and ALK4 receptor dimer for myostatin) can lead to a decrease in the expression of the muscle fiber hypertrophic factor IGF-1 [93, 183].
TGFβ1 is expressed by multiple different cell types such as FAPs/fibroblasts and endothelial cells, but mainly by macrophages: over 75%of these cells in the mdx diaphragm express TGFβ [90, 184]. One important feature of TGFβ1 signaling is that ligand gene expression may not directly lead to downstream signaling activation. This has been demonstrated in animal models where TGFβ expression levels do not correlate with the amount of fibrosis observed [185, 186]. Indeed, secreted TGFβ1 is often found bound to LTBP and is stabilized but kept inactive until LTBP cleavage [187]. Notably, DMD muscles exhibit elevated LTBP4, regulating TGFβ1 availability [188].
Treating mdx mice with Nilotinib (which inhibits p38-like kinases downstream of TGFβ) rescues the dystrophic phenotype by decreasing FAP numbers and the associated fibrosis [56, 59]. The dystrophic environment alters the effect of macrophages toward FAPs. Inflammatory macrophages (Ly-6C+CX3CR1low) isolated from fibrotic mdx muscle have lost their ability to induce fibroblast apoptosis and instead stimulate collagen I expression [56]. This effect is reversed by anti-TGFβ antibody treatment. Moreover, inflammatory macrophages from fibrotic mdx muscle express more LTBP4, which allows for more latent TGFβ1 to be stabilized within the ECM compared to non-fibrotic mdx muscle. In mdx muscle, FAPs secrete MMP14 and BMP1 proteases to release TGFβ1 from LTBP4 [56].
Treatment of mdx mice with the AMPK activating compound metformin (otherwise used for the treatment of type 2 diabetes) promotes pro-inflammatory to pro-regenerative macrophage phenotype skewing, reduces fibrosis, and improves muscle morphology [56]. In mdx:IL-10-KO mice, macrophages increase their expression of pro-inflammatory cytokines and an increase in mature TGFβ and collagen I secretion within muscle tissue is observed [189]. Similarly, fibrinogen (a soluble protein released into the blood in response to stress which accumulates in mdx and DMD muscles)-activated BMDMs treated with blocking IL-1β antibodies exhibit a decrease in Tgfb gene expression. On the other hand, mdx fibroblasts treated with conditioned medium of fibrinogen activated BMDM increases collagen I (Col1a1) expression, and this pro-fibrotic effect can be reverted with TGFβ1 blocking antibody [190]. Together, these studies demonstrate that the atypical pro-inflammatory macrophages found in dystrophic muscle not only act on myofiber damage and repair, but are also capable of directly inducing fibrosis. Thus, in dystrophic conditions, the dysregulation of macrophage phenotype induces a vicious cycle between macrophage LTBP4-TGFβ expression, FAP survival and ECM component expression that leads to fibrosis.
It should be noted that the effects of TGFβ are not limited to FAP/fibroblasts. SCs and endothelial cells also express receptors for this molecule. In vitro treatment of SCs and endothelial cells with TGFβ results in decreased myotube and angiotube formation and is associated with an increase in fibrotic gene expression [174]. By using specific endothelial cell and SC tracking mice, this phenomenon has been replicated in vivo in 6-month-old mdx mice. Specifically, approximately 12%of SCs downregulated a myogenic fate marker (loss of α7-integrin) and showed elevated Col1a1 and fibronectin extra domain A (Eda-Fn1) expression. Similarly, 30%of the original endothelial cell population reduced CD31 expression, with a concomitant increase in Col1a1 and Eda-Fn1. Within dystrophic muscle, infiltrating macrophages also become pro-fibrotic expressing more collagen I and less CD45. These “fibrotic” SCs, endothelial cells and macrophages represent only 1-2%of the total fibrogenic population, and while they may not have a major impact on fibrosis per se, they may no longer be capable of participating in muscle and vessels formation. Indeed, myogenic cells, endothelial cells and macrophages must communicate for effective muscle regeneration and it has been observed that mdx mice also exhibit impaired vessels formation and functional vascular defects [98, 192].
Other proteins expressed by macrophages that modulate MD progression
Several studies have pointed out additional proteins expressed by macrophages that exacerbate or attenuate muscular dystrophy progression.
TREATMENT OF MUSCULAR DYSTROPHIES: WHAT ABOUT MACROPHAGES?
Excessive inflammation within dystrophic muscle is demonstrably more deleterious than beneficial. The only treatment that has shown a delay in disease progression is the use of glucocorticoids. Unfortunately, these potent anti-inflammatory drugs have significant side effects. Prednisone, one of the most widely used glucocorticoids, decreases inflammation and delays the progression of DMD, prolonging ambulation and modestly improving muscle strength and cardiopulmonary function. Side effects include bone fragility, weight gain, mood changes, and even muscle weakness [220–224]. Glucocorticoids stimulate the AKT1/FOXO1 pathway, which decreases protein synthesis and increases protein catabolism and is responsible for the seemingly contradictory muscle weakness and atrophy observed in patients treated with this drug [225]. Another potent glucocorticoid is Dexamethasone, but side effects are severe, making it an unappealing candidate for long-term treatment. Deflazacort is a less potent glucocorticoid that has a similar effect to prednisone but with a reduced number of side effects [224, 226]. In the end, a combination of the different drugs seems the most appropriate way to delay MD progression [227]. The main concern stays the poor knowledge of long-term effects. For example, mdx mice treated with prednisone for 50 days showed an improvement in early disease progression, which was subsequently lost when treatment was continued to 100 or 150 days [221]. In addition, whether the effects of corticosteroids are mainly through abatement of inflammation or though one of the other pleiotropic effects of these compounds is not yet clear.
Therapeutic approaches that harness macrophages are beginning to emerge. 24 hours after an acute ischemia/reperfusion injury, intra-muscular injection of pro-inflammatory macrophages has been shown to improve muscle regeneration, characterized by increased muscle force, myofiber diameter and by decreasing collagen deposition at 14 days post-reperfusion [228]. At 5 and 7 days after reperfusion, a decrease in damaged muscle area is observed (probably via improved removal of dead cells). While the total number of macrophages was unchanged between control and the macrophage-injected muscles, an increase in the number of CD206+ macrophages was observed 5 days post-perfusion in the macrophage-injected muscle, demonstrating that the injected pro-inflammatory macrophages switched toward a pro-regenerative phenotype within the treated muscle [228]. Injection of human macrophages into damaged muscle of immuno-suppressed mice, together with human myoblasts, improved myoblast proliferation and led to better host cell integration within the myofibers [229]. As observed in mice, five days after their injection pro-inflammatory human macrophages expressed anti-inflammatory markers, further demonstrating their capacity to locally change phenotype during progression of muscle regeneration [229]. Of note, one of the main causes of failure of cell therapy for MDs is the poor survival and migration capacity of SCs and myoblasts after intramuscular injection. Co-injection of BMDM and SCs into mdx muscle increases their proliferation, survival and migration [51]. Thus, “non-dystrophic” macrophages seem to support injected myoblasts in their regeneration of muscle tissue. Modification of macrophage phenotype could be beneficial to dystrophic muscle not only because macrophages act negatively on fibrogenic cells within the progression of MD, but also because of the evidence supporting their use in these cell therapies. Interestingly, Novak et al. demonstrated that myoblasts are not the only cells capable of delivering phosphorodiamidate morpholino oligomers to myofibers, and that macrophages are also potent releasers of these therapeutics, making them an attractive candidate for in situ delivery to myoblasts and myofibers [230].
CONCLUSION
Macrophages have been studied for decades and most of their functions are now understood. Indeed, their role beyond infection response, specifically as tissue resident cells involved in the remodelling of tissues (development, regeneration and repair) are well understood within the research community. However, the manipulation of their inflammatory state in order to direct their trophic functions toward tissue-resident cells is far from being defined. MDs, and especially DMD are multifactorial diseases where necrosis, chronic inflammation, defects in angiogenesis, fibrofatty infiltration, and tissue remodelling occur asynchronously within the tissue. The defect in macrophage function in these diseases could be one of the reasons for poor outcome of cell and gene therapies. It is therefore important that further efforts be made to safely manipulate macrophage dynamics so that they might be used to therapeutic effect as part of a MD rescue approach. Today, single cell technology such as CITE-seq (Cellular Indexing of Transcriptomes and Epitopes by Sequencing) should allow the community to link macrophage function, polarization state and gene expression, to find appropriate therapeutic gene and protein targets. We imagine a future where “re-booting” or resynchronizing the inflammatory system would allow improvements to the muscle repair cycle, by delaying fibrosis apparition, and the loss of muscle function; or as synergistic tools used alongside gene and cell therapies to improve their efficacy.
Footnotes
ACKNOWLEDGMENTS
The authors would like to thank Dr. Morten Ritso and Mark Hamer for their helpful suggestions. Figures were created using BioRender.com
AUTHORS’ CONTRIBUTIONS
M.S. and M.T. drafted the review. G.M and F.M.V.R. revised and reviewed the manuscript. All authors read and approved the final manuscript.
FUNDING
The authors were supported by European Community (ERCStG2011, RegeneratioNfix 280611) and the Association Française contre les Myopathies (AFM, 20002) to MS and GM, by Fondation pour la Recherche Médicale (FRM, 40248), by the European Molecular Biology Organization (EMBO, ALTF 115-2016), by the French Association Française contre les myopathies (AFM, 22576), and by Michael Smith Foundation for Health Research (MSFHR, 18351) to M.T.; and by the Canadian Institutes of Health Research (CIHR-FDN-159908) to F.M.V.R.
COMPETING INTERESTS
The authors declare that they have no competing financial interests. The funding agencies had no role in the review’s design, the decision to publish, or preparation of the manuscript.