
Abstract
Background:
Leigh syndrome (LS) is the most frequent paediatric clinical presentation of mitochondrial disease. The clinical phenotype of LS is highly heterogeneous. Though historically the treatment for LS is largely supportive, new treatments are on the horizon. Due to the rarity of LS, large-scale interventional studies are scarce, limiting dissemination of information of therapeutic options to the wider scientific and clinical community.
Objective:
We conducted a systematic review of pharmacological therapies of LS following the guidelines for FAIR-compliant datasets.
Methods:
We searched for interventional studies within Clincialtrials.gov and European Clinical trials databases. Randomised controlled trials, observational studies, case reports and case series formed part of a wider MEDLINE search.
Results:
Of the 1,193 studies initially identified, 157 met our inclusion criteria, of which 104 were carried over into our final analysis. Treatments for LS included very few interventional trials using EPI-743 and cysteamine bitartrate. Wider literature searches identified case series and reports of treatments repleting glutathione stores, reduction of oxidative stress and restoration of oxidative phosphorylation.
Conclusions:
Though interventional randomised controlled trials have begun for LS, the majority of evidence remains in case reports and case series for a number of treatable genes, encoding cofactors or transporter proteins of the mitochondria. Our findings will form part of the international expert-led Solve-RD efforts to assist clinicians initiating treatments in patients with treatable variants of LS.
INTRODUCTION
Mitochondrial diseases are common inherited metabolic disorders, of which the commonest is Leigh syndrome (LS) [1, 2]. The clinical phenotype of LS is comprised of neurological symptoms with basal ganglia and/or brainstem dysfunction. The diagnostic criteria include clinical and radiological features and biochemical evidence of abnormal energy metabolism due to defects in the oxidative phosphorylation (OXPHOS) pathway or pyruvate dehydrogenase complex (PDHc). If the diagnostic criteria are not fulfilled, the condition can be classified as Leigh-like syndrome [2]. LS usually presents in early childhood. with developmental delay, encephalopathy, dystonia and symmetrical T2 hyperintensities in the basal ganglia and brainstem, but some cases with late-onset are known. Additionally, systemic issues outside of the central nervous system such as renal failure, cardiomyopathy, diabetes mellitus can be present, making LS a disease with diverse clinical features [3, 4].
Despite well-defined diagnostic criteria, the combination of clinical heterogeneity and rarity can lead to delay in diagnosis.Whilst there are both complete and ongoing clinical trials in LS [5], the lack of clearly-presented evidence and vast amount of literature, the majority of which lies in case reports and case series, prevent timely instigation of treatment by clinicians. Moreover, there is a lack of guidance regarding which of the over 75 causative genes and which biochemical pathways in LS [2] are treatable, without extensive literature searching. The combination of diagnostic delay and lack of systematic evaluation of treatments inevitably impacts the clinical course of LS and the likelihood of patient response to treatment.
The RD-Connect Genome-Phenome Analysis Platform (GPAP) is an online tool for diagnosis and gene discovery in rare disease research (https://rd-connect.eu/what-we-do/omics/gpap/). As part of the H2020 research project Solve-RD rare disease experts aim towards creating a database of treatable rare disease, the ‘Treatabolome’, at gene and variant levels [6]. Previous Treatabolomes have been created for congenital myasthenic syndromes [7], inherited neuropathies [8] and laminopathies [9]. As part of this international effort, we have systematically reviewed existing evidence of pharmacological treatments of LS.
METHODS
Systematic review protocol
We aimed to identify any gene/variant specific pharmacological treatments in LS and assessed the strength of the associated supporting evidence as part of the creation of a LS ‘Treatabolome’. The systematic review was designed following the guidelines set out by Atalaia et al. 2020 for creating a Treatabolome with FAIR-compliant datasets [6].
Literature search strategy
The conditions studied were genetically confirmed LS as previously described, including clinical, radiological criteria and known LS-associated genetic variants [2]. We searched electronic databases MEDLINE via PubMed, ClinicalTrials.gov and the European Clinical Trials Register. A full search strategy including search terms and filters used for each database is described in Supplementary File 1.
We identified 1,056 articles from PubMed, 12 clinical trials from ClinicalTrials.gov and 2 from European Clinical Trials Register (Fig. 1). Relevant reviews and other secondary literature identified from PubMed were flagged and screened for relevant primary research that met the inclusion criteria. This identified an additional 123 papers. All studies were uploaded onto the Rayyan QCRI platform for subsequent screening and removal of duplicates (Fig. 1).
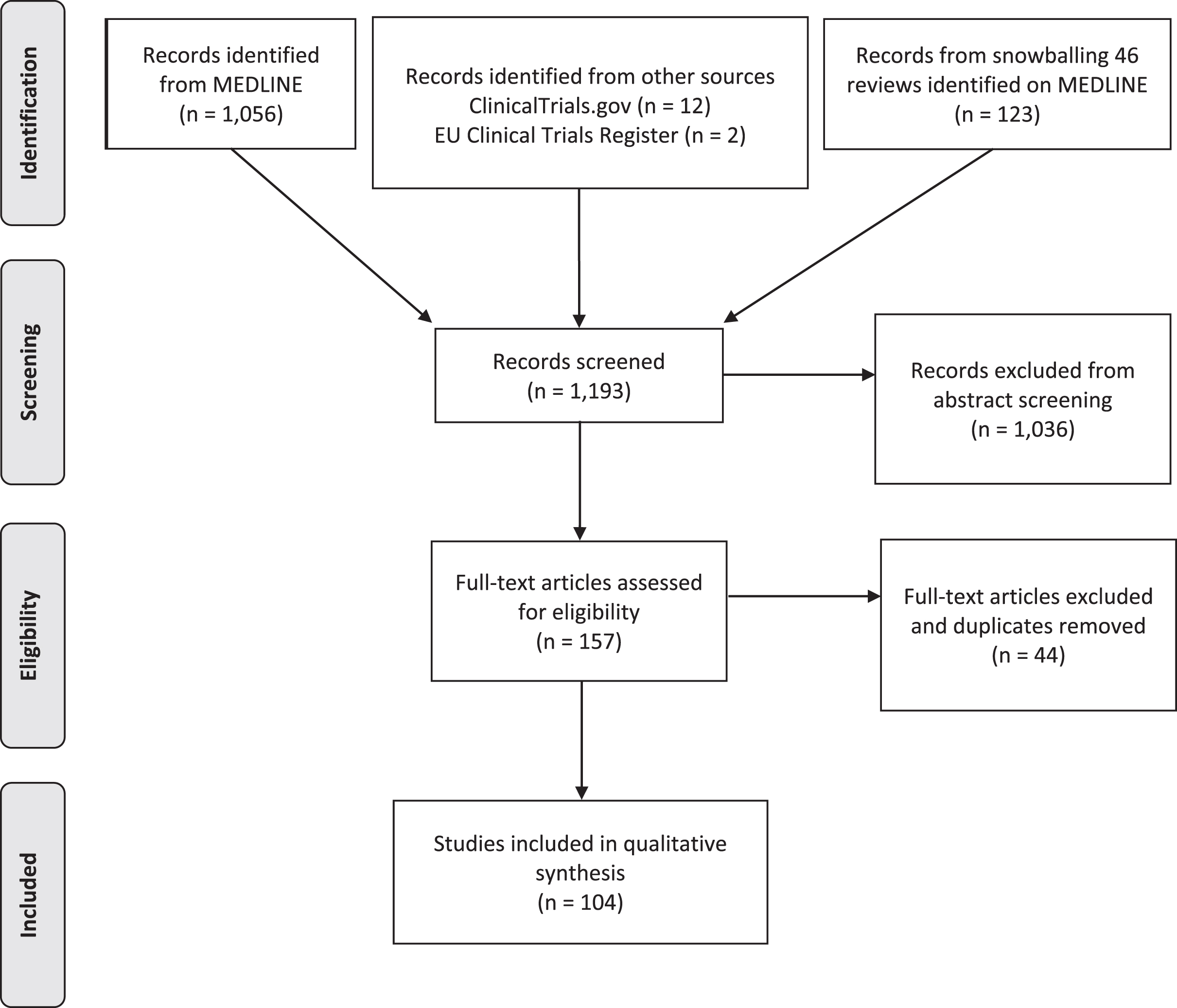
PRISMA flow diagram of articles screened for treatments in Leigh syndrome.
Inclusion & exclusion criteria for screening
Following compilation of all the papers identified from each database, title & abstract screening was undertaken by 2 independent blinded reviewers (ZL, MYT) and arbitrated by an expert reviewer (RH). The following inclusion and exclusion criteria were implemented for both title and abstract screening and full-text screening (Fig. 2).

Inclusion and exclusion criteria for screening Leigh syndrome treatments.
Data extraction
For the 104 papers that met our inclusion criteria, all relevant information was extracted onto a standardised data form as described in the guidelines [6] (Fig. 3). In studies with multiple patients on different treatment regimes, individual gene variants and all treatments trialled for each individual patient were recorded where such information was available. Each paper was evaluated by 2 independent blinded reviewers (ZL, MYT) using both the OCEBM 2011 Levels of Evidence score [10] and the Jadad score [11]. Discrepancies in scoring were resolved by a third reviewer (RH).

Data extraction from articles discussing treatment in Leigh syndrome to develop a FAIR-compliant dataset.
RESULTS
In total, 1,193 records were screened from PubMed, EU Clinical Trials Register, Clinicaltrials.gov and review articles. A total of 157 papers passed the inclusion criteria. After removing duplications, 104 studies were included in our analysis. The majority of evidence remained in case reports or case series scoring 4 (44.7%, n = 46) or 5 (50.5%, n = 52) on OCEBM and 0 (95.1%, n = 98) on Jadad scoring (Fig. 4). The highest levels of evidence were associated with completed clinical trials in only 3 compounds: Vatiquinone (EPI-743) (n = 2) KH176 (n = 1) and cysteamine bitartrate (RP103) (n = 2), but were all open-labelled studies (level 3 OCEBM, Jadad 1). Randomized multi-centre clinical trials with KH176 are ongoing, and a further trial with novel compound Nabsirolimus (ABI-009) is scheduled to begin recruitment late 2021 (Fig. 5).
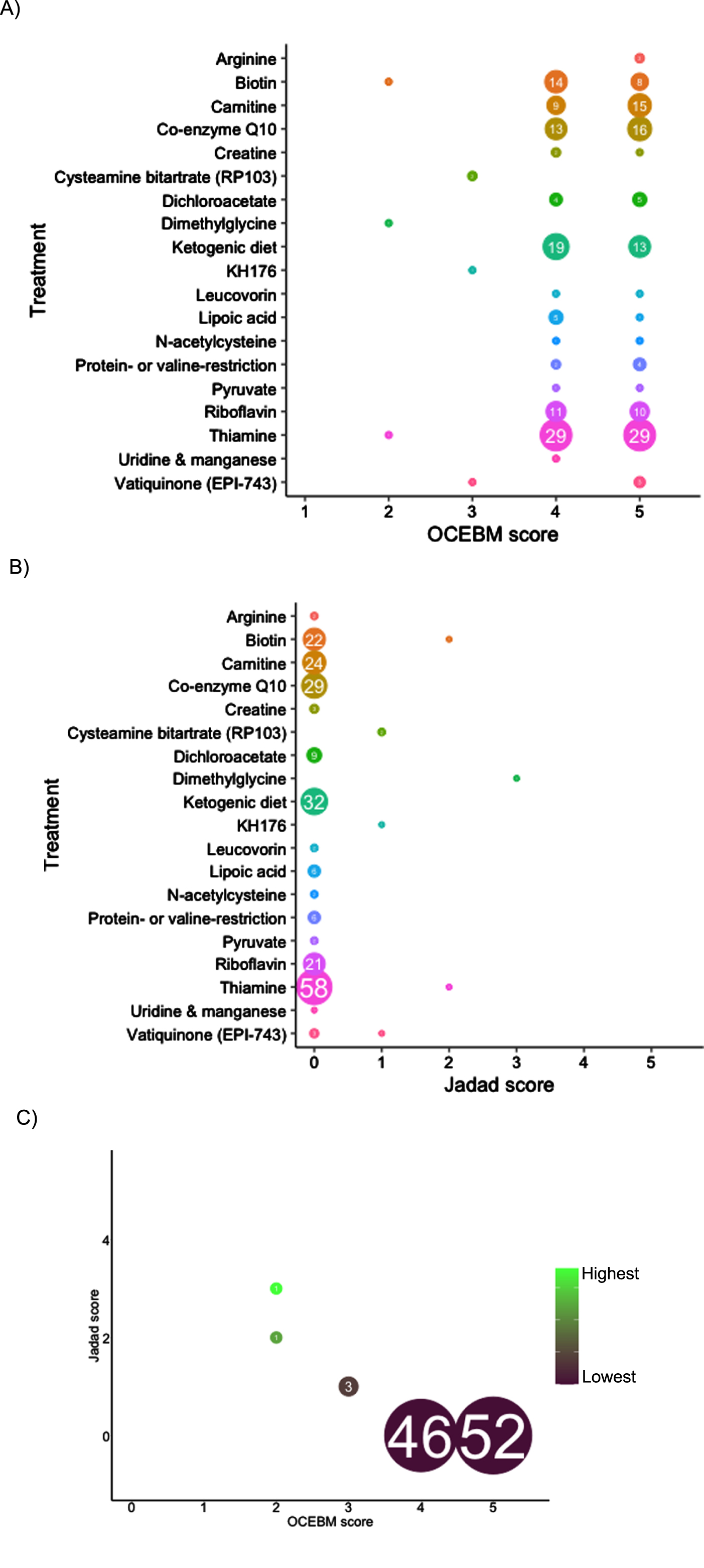
Level of evidence of treatment in Leigh syndrome according to treatment, number of studies and OCEBM score (4A), Jadad score (4B), combined Jadad and OCEBM score.
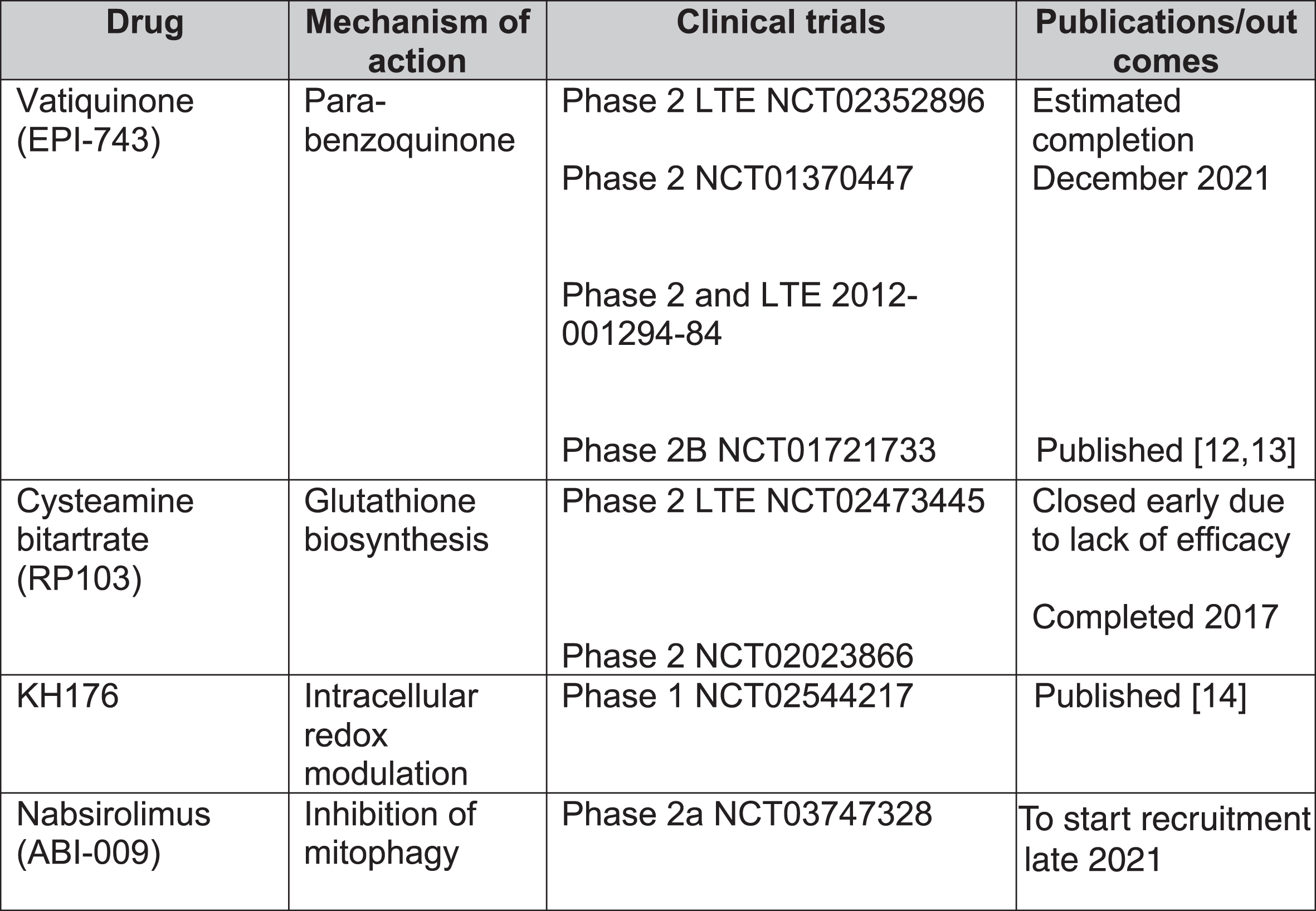
Clinical trials for treatment of Leigh syndrome. LTE = long-term extension.
EPI-743
EPI-743, a novel para-benzoquinone that repletes intracellular gluthathione more potently than co-enzyme Q10 or idebenone, has been tested in open-label clinical trials in genetically confirmed inherited respiratory chain diseases [13]. Enns et al. reported that 11 of the 12 participants showed clinical improvement and increased regional and whole brain technetium-99m-hexamethylpropylene oxime (HMPAO) uptake on single photon emission computed tomography (SPECT) [12]. In a phase 2a single-site clinical trial, Martinelli et al. reported significant improvement of the Newcastle Paediatric Mitochondrial Disease Scale (NPMDS), Gross Motor Function Measure and PedsQL Neuromuscular Module in 10 children with genetically-confirmed Leigh syndrome [13].
Cysteamine bitartrate
Cysteamine is required for gluthathione biosynthesis to reduce oxidative stress levels and has been used for lysosomal storage disease cystinosis [15]. So far, there have been 2 open-labelled studies (NCT02473445 and NCT02023866) on various mitochondrial diseases. The phase 2 long-term extension study was terminated early due to lack of efficacy.
KH176
KH176 is a redox-modulating agent. A phase 1 study (NCT02544217) for various mitochondrial diseases, including LS, reported that the agent was well-tolerated but caused QTc prolongation and T-wave morphological changes at higher doses [14]. Randomized multi-centre phase 2 studies are currently recruiting including mitochondrial disorders such as MELAS, MIDD, LS and mitochondrial myopathies as well as mitochondrial encephalopathies. A safety and efficacy trial has been completed in m.3243A > G-associated mitochondrial disease [16].
Nabsirolimus (ABI-009)
Nabsirolimus is an albumin-bound sirolimus nanoparticle suspension which inhibits mitophagy [5]. A phase 2, open-label study for LS patients with moderate severity according to NPMDS score plans to begin recruitment of patients in August 2021 (NCT03747328).
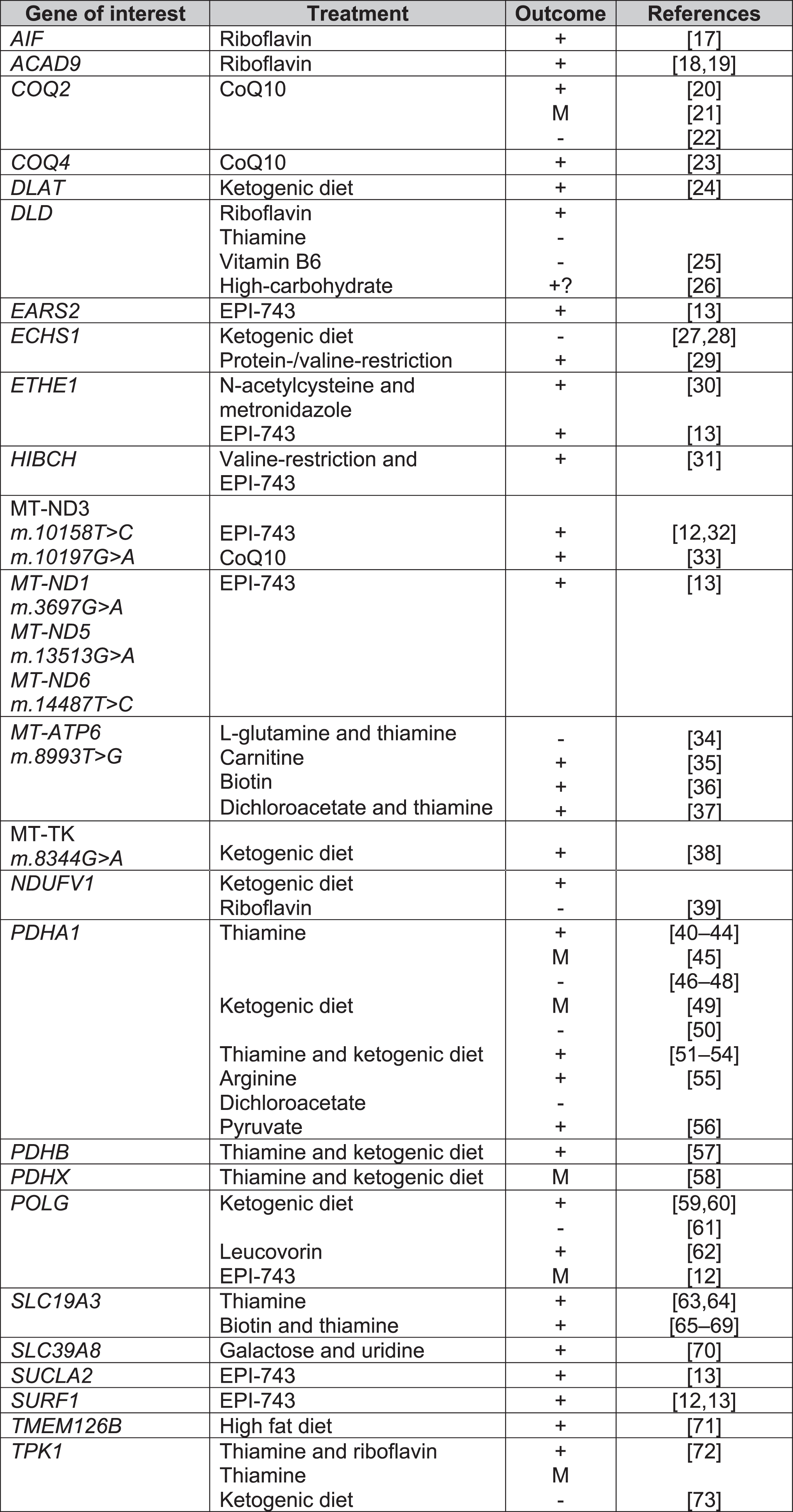
Treatments for Leigh syndrome according to causal gene. Key: + = overall positive outcome, M = mixed outcome, - = negative outcome, CoQ10 = co-enzyme Q10, EPI-743 = Vatiquinone.
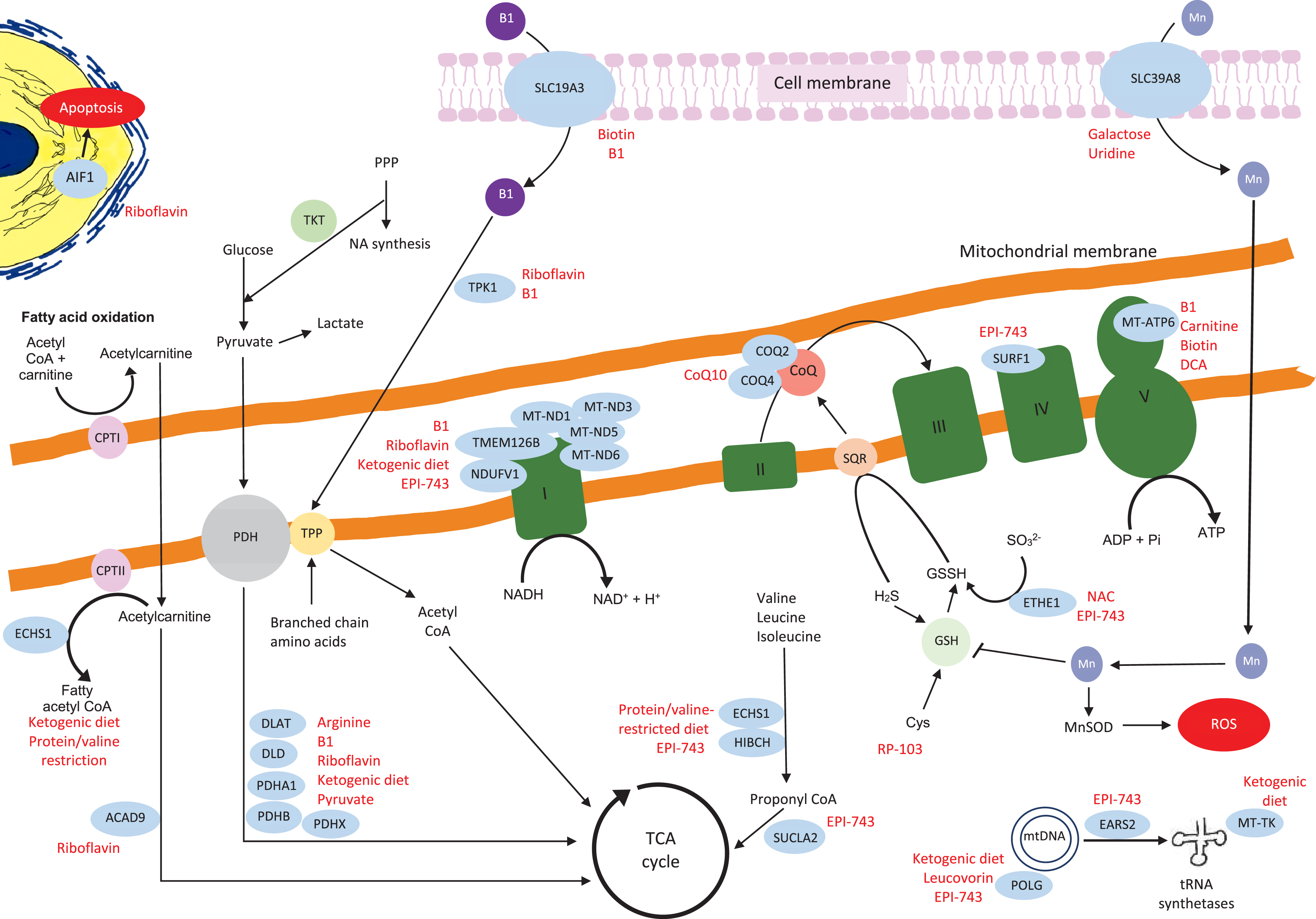
Schematic representation of the mitochondrial genes and proteins with their links to the other metabolic pathways. The genes where we identified relevant references on treatments are in blue circles, the potential treatments are highlighted in red. The details of the relevant treatments are explained above. Key:B1 = thiamine, TPP = thiamine pyrophosphate, TKT = transketolase, DCA = dichloroacetic acid, PPP = pentose phosphate pathway, NA = nucleic acid, PDH = pyruvate dehydrogenase, CPT1 = carnitine palmitoyltransferaseI, Actyl CoA = acetyl co-enzyme A, CoQ = coenzyme Q, TCA = tricarboxylic acid, H2S = hydrogen sulfide, GSH = glutathione, GSSH = glutathione persulfide, SQR = sulfide:quinone oxidoreductase, Cys = cysteine, Mn = manganese, MnSOD = manganese-superoxide dismutase, ROS = reactive oxygen species.
Biotin and thiamine
The SLC19A3 gene encodes a thiamine transporter. SLC19A3 mutations can lead to Biotin-Responsive Basal Ganglia Disease (BBGD). A total of 22 and 57 studies included biotin and thiamine supplementation respectively, most commonly in combination as a “mitochondrial cocktail” of treatments. Only 2 case series tested biotin or thiamine monotherapy versus biotin and thiamine combined treatment. Debs et al. reported clinical and radiological improvement in both biotin monotherapy and combination therapy in 2 siblings with missense mutations in SLC19A3 [74], whilst Tabarki et al. reported no significant difference in Burke-Fahn-Marsden Dystonia Rating Scale (BFMDRS) following thiamine monotherapy or combination therapy in an open-labelled prospective study in 20 children with SLC19A3 c.1264A > G (p.Thr422Ala) mutation. There was, however, significantly quicker recovery from acute crises (2 days versus 3 days) with combination therapy [65].
Delays in treatment initiation can limit meaningful clinical outcome. Yamada et al. and Kevelam et al. reported no improvement with biotin therapy in patients with advanced disease [75, 76]. Kevelam et al. initiated biotin therapy for a patient with homozygous SLC19A3. The treatment was initiated age 19 months but there was evidence of severe cortical atrophy on MRI age 14 months [75].
Co-enzyme Q10 (ubiquinone)
Co-enzyme Q10 (CoQ10) is required for oxidative phosphorylation in mitochondria, acting as an electron carrier between complex I, II and III [77]. A large number of studies were excluded for data extraction due to secondary CoQ10 deficiency or patients not fulfilling the diagnostic criteria for LS clinically, radiologically or genetically. As with biotin and thiamine, in the majority of the 28 studies which included CoQ10, CoQ10 formed part of a mitochondrial cocktail of supplements for LS patients (Supplementary Figure 2). Six studies assessed CoQ10 alone, of which 4 reported improvement in patients with mutations in m.10197G > A, COQ2, COQ4 [20, 33]. It was noted that clinical deterioration occurred if patients were already in late-stages of disease before treatment initiation [21]. Improvement in renal function were reported with CoQ10 initiation [20]; however, Scalais et al. reported a child who developed massive proteinuria despite CoQ10 supplementation in infancy [22].
Pyruvate
Pyruvate provides NAD+ restoring ATP production and reducing the NADH/NAD + ratio from oxidative phosphorylation disorders [78, 79]. The majority of studies included biochemical pyruvate dehydrogenase deficiencies which were not radiologically or genetically-confirmed LS. Only 2 papers passed all inclusion and exclusion criteria. The highest level of evidence was a case series by Fuji et al. including 2 genetically confirmed LS patients with the following variants m.8993T > G and m.9176 T > C, who were bedbound but had improvements in the Newcastle Paediatric Mitochondrial Disease Scale (NPMDS) after pyruvate treatment [79].
Dichloroacetic acid (DCA) inhibits pyruvate dehydrogenase kinase, which in turn inhibits pyruvate dehydrogenase [80]. Our study confirms the mixed clinical outcomes from DCA. DCA can lead to worsening peripheral neuropathy, even with prophylactic administration of thiamine [81]. Successful improvement of midbrain hyperintensities with DCA and thiamine therapy in a patient with MT-ATP6 m.8993T > C were reported by Fujii et al. [37]. Conversely, Koga et al. reported a child with a PDHE1 α mutation who deteriorated with DCA but responded to pyruvate therapy [56].
Riboflavin
Flavin adenine dinucleotide (FAD) precursor protein, riboflavin, was used in 18 studies, but the majority were part of a mitochondrial cocktail treatment. A total of 3 studies reported riboflavin monotherapy. Two studies reported improvement in patients with ACAD9 mutations [18, 19]. Gerards et al. reported restoration of complex I activity in a family with homozygous ACAD9 (c.1594C > T) mutation [19]. Ghezzi et al. described 2 children born from monozygotic twin sisters with mutations in AIF1, 1 patient received treatment with riboflavin with initial improvement but later deteriorated with further seizures and tetraplegia [17].
Carnitine
Carnitine is involved in fatty acid oxidation [82] and is used as a neutraceutical (mitochondrial cocktail) with other co-factors and antioxidants to bypass the electron transport chain [83]. Carnitine featured as a mitochondrial cocktail treatment in case series or case reports but often with no effect [28, 85]. A case report by Toth et al. reported a patient with m.8993T > C mutation acutely losing ambulation with an upper respiratory tract infection. It is unclear if the clinical improvement with subsequent carnitine supplementation coincided with resolution of their respiratory tract infection. The authors reported low plasma and muscle carnitine levels prior to treatment. Further deterioration in ataxia and weakness was noted at the age of 13 and then improvement with adjusted dosing of carnitine according to weight [35].
N-acetylcysteine
N-acetylcysteine (NAC) is a precursor of antioxidant agent glutathione which binds toxic sulfide [15, 30]. We found 2 studies assessing the effectiveness of NAC treatment. Most notably, Viscomi et al. described improvement in seizure-frequency, neurological improvement, reduction of acrocyanosis and petechiae in 5 children with homozygous 505 + 1G > T splice-site mutations in ETHE1. The patients also had improvements in diarrhoea, which may be explained with the combination therapy with bactericidal and prokinetic agent Metronidazole [30]. Shayota et al. reported developmental improvement with NAC in a patient with ECSH1 mutation; however, the patient also received a valine-restricted diet [29].
Other supplements
Isolated studies have been published for arginine [55], leucovorin (folinic acid) [62], uridine and galactose [70] stating a positive effect. Manganese deficiency in SLC39A8 variants leads to the decrease in manganese superoxide dismutase and oxidative stress [86]. Treatment with uridine and galactose in SLC39A8 is reported to improve transferrin glycosylation pattern [70]. Further studies are required to fully assess the effectiveness of such treatments.
Diets
Dietary modifications are known for use of epilepsy-control. Ketogenic diets are thought to promote fatty acid β-oxidation as an alternative pathway to oxidative phosphorylation [39]. Effectiveness in epilepsy control, eye movements and mental development has been reported in ECHS1 [28], POLG [60], TMEM126B [71], PDHA1 leading to pyruvate dehydrogenase deficiency [43].
Protein and valine-restriction have been reported to be effective in patients with mutations involved in valine-degradation, such as HIBCH [31] and ECSH1 [29]. It is believed that such diets help to circumvent the accumulation of toxic metabolites such as methacrylyl-CoA and acryloyl-CoA [29]. However, ketogenic diet can induce metabolic acidosis and lead to clinical dete- rioration in LS [73]. To date, the evidence of dietary modifications is limited to case reports and small case series.
Measurable outcomes
The majority of treatments for LS are from low quality studies. Clinical improvements reported in case reports and case series are often without quantitative clinical scales. Although case series rank as low quality of evidence, we note that some studies used clinically-validated disease scores such as NPMDS [12, 13] and BFMDRS [65].
Promising biomarkers are emerging in LS, such as glutathione status as a marker of redox for EPI-743 therapy [87]. Radiological features such as basal ganglia lesions [74] and HMPAO on SPECT [12] are associated with clinical improvement in LS and could serve as potential therapeutic biomarkers.
CONCLUSIONS
We have conducted a systematic review of mutation-specific treatments in LS according to FAIR-principles, as part of an international effort to develop a rare disease Treatabolome. The majority of evidence of LS treatments relies on case reports and case series, resulting in publication bias due to the tendency towards reporting treatment-responsive cases. However, these reports should not be overlooked as a number of LS genetic variants are highly responsive to treatments. This holds promise for the development of future therapies and clinical trials. Clinicians with patients of these treatable genotypes may wish to consider experimental use of these therapies with the guidance of specialised mitochondrial services. There are a small number of clinical trials which are still actively recruiting and EPI-743 has reported clinical improvement in LS patients.
Clinical heterogeneity may affect trial outcomes. However, clinical trials may not be needed when the clinical response and disease mechanism is clear; an example would be BBGD, where prospective case series compared biotin/thiamine monotherapy or combination therapies using validated clinical outcome measures [65]. The LS Treatabolome will allow clinicians to appropriately assess the existing evidence for treatable LS genotypes. Due to the clinical heterogeneity of LS, we propose that MRI features and a genetically confirmed variant to be used as part of the selection criteria for future clinical trials. However, due to the relative low prevalence of LS, it is likely that future trials will continue to incorporate this patient group within other mitochondrial disorders.
Treatment-responsiveness, as documented in several trials and case reports, relies on early recognition and intervention. To that end, our findings will form part of the international expert-led Solve-RD efforts to assist clinicians initiating treatments in patients with treatable-variants of LS.
Footnotes
ACKNOWLEDGEMENTS
M.Y.T. was supported by the Addenbrookes Charitable Trust (G104114). R.H. is a Wellcome Trust Investigator (109915/Z/15/Z), who receives support from the Medical Research Council (UK) (MR/N025431/1 and MR/V009346/1), the European Research Council (309548), the Newton Fund (UK/Turkey, MR/N027302/1), the Addenbrookes Charitable Trust (G100142), the Evelyn Trust, the Stoneygate Trust, the Lily Foundation and an MRC strategic award to establish an International Centre for Genomic Medicine in Neuromuscular Diseases (ICGNMD) MR/S005021/1. The study was further supported by the Horizon 2020 research and innovation program via grant 779257 “Solve-RD”. This research was supported by the NIHR Cambridge Biomedical Research Centre (BRC-1215-20014). The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to report.