
Abstract
Background:
Dominant and recessive autosomal pathogenic variants in the three major genes (COL6A1-A2-A3) encoding the extracellular matrix protein collagen VI underlie a group of myopathies ranging from early-onset severe conditions (Ullrich congenital muscular dystrophy) to milder forms maintaining independent ambulation (Bethlem myopathy). Diagnosis is based on the combination of clinical presentation, muscle MRI, muscle biopsy, analysis of collagen VI secretion, and COL6A1-A2-A3 genetic analysis, the interpretation of which can be challenging.
Objective:
To refine the phenotypical spectrum associated with the frequent COL6A3 missense variant c.7447A>G (p.Lys2483Glu).
Methods:
We report the clinical and molecular findings in 16 patients: 12 patients carrying this variant in compound heterozygosity with another COL6A3 variant, and four homozygous patients.
Results:
Patients carrying this variant in compound heterozygosity with a truncating COL6A3 variant exhibit a phenotype consistent with COL6-related myopathies (COL6-RM), with joint contractures, proximal weakness and skin abnormalities. All remain ambulant in adulthood and only three have mild respiratory involvement. Most show typical muscle MRI findings. In five patients, reduced collagen VI secretion was observed in skin fibroblasts cultures. All tested parents were unaffected heterozygous carriers. Conversely, two out of four homozygous patients did not present with the classical COL6-RM clinical and imaging findings. Collagen VI immunolabelling on cultured fibroblasts revealed rather normal secretion in one and reduced secretion in another. Muscle biopsy from one homozygous patient showed myofibrillar disorganization and rimmed vacuoles.
Conclusions:
In light of our results, we postulate that the COL6A3 variant c.7447A>G may act as a modulator of the clinical phenotype. Thus, in patients with a typical COL6-RM phenotype, a second variant must be thoroughly searched for, while for patients with atypical phenotypes further investigations should be conducted to exclude alternative causes. This works expands the clinical and molecular spectrum of COLVI-related myopathies.
Keywords
INTRODUCTION
Collagen VI is an extracellular matrix protein pre-sent in most tissues, notably in muscle, skin, tendon and blood vessels. Dominant or recessive autosomal pathogenic variants in each of the three “major” genes encoding the collagen VI α-chains (COL6A1, COL6A2, COL6A3) underlie collagen VI-related myopathies (COL6-RM), a heterogeneous group of disorders marked by a combined muscle and connective tissue involvement including joint laxity and contractures, as well as characteristic cutaneous abnormalities (i.e. keloids, keratosis pilaris and soft/velvety skin) in addition to muscle weakness.
COL6-RM clinical spectrum ranges from early-onset severe conditions (Ullrich congenital muscular dystrophy, UCMD) through phenotypes of intermediate severity to milder forms (Bethlem myopathy, BM) [1, 2]. Classically, UCMD patients present with congenital weakness and hypotonia, delayed motor milestones, associated with proximal joint contractures and concomitant marked distal hyperlaxity. Rigid spine, scoliosis, hip dislocation/dysplasia and prominent calcaneus are common features. Progressive weakness leads to early loss of ambulation in most patients, and restrictive respiratory involvement occ-urs in most severely affected patients during the first two decades of life [3]. Conversely, BM phe-notype is marked by milder proximal weakness ass-ociated with contractures typically affecting Achilles tendons, elbows, pectoralis, long finger flexors and interphalangeal joints. Although a slowly-progres-sive condition, two-thirds of patients over the age of 60 years may need assistance with ambulation [4, 5].
Nonetheless, there is a wide clinical variability, and intermediate phenotypes are now well recognized. Interestingly, patients can present with predominan-tly proximal weakness and very few or absent contra-ctures or distal hyperlaxity, more akin to a limb-girdle muscular dystrophy (LGMD) [6, 7]. Furthermore, recessive mutations in COL6A2 have also been associated with severe and widespread contractures known as myosclerosis [8].
Diagnosis is based on the combination of clinical presentation supported by muscle MRI, muscle biopsy findings, immunohistochemical examination of collagen VI secretion and analysis of the COL6A1-3 genes sequences. Characteristic muscle MRI findings of COL6-RM include a peculiar fatty replacement starting around the fascia surrounding the muscle (“outside-in” picture) with the presence of the so-called “central-cloud or shadow” typically affecting the rectus femoris [9].
Although UCMD was initially described as an autosomal recessive condition, dominant, mostly de novo, mutations have also been identified. Conversely, rare autosomal recessive pathogenic variants have been reported in BM patients, although it is mostly inherited as a dominant condition [4, 10]. Interpretation of genetic variants can be challenging, since there are few hotspots, and the COL6A1-3 genes are highly polymorphic. Furthermore, the clinical consequence of the many COL6A1-3 variants that affect residues of unknown impact on the heterotrimeric assembly of COLVI (in particular variants outside of the triple helical domain) may be difficult to interpret and to validate experimentally.
We report a total of 16 patients carrying the COL6A3 missense variant c.7447A>G either as compound heterozygotes with another COL6A3 variant (12 patients from 10 families) or in a homozygous state (4 unrelated patients). Upon extensive analysis of the clinical phenotype and ancillary tests including muscle imaging pattern, muscle biopsy histology and COLVI secretion in dermal fibroblasts, we discuss the potential pathogenicity of this variant.
MATERIAL AND METHODS
Patients
Through an international collaboration, we identified 16 patients from 14 families (Table 1) carrying the c.7447A>G variant in the COL6A3 gene in heterozygosity with another COL6A3 mutation (n = 12) or in homozygosity (n = 4). Clinical data and ancillary tests (including serum CK levels, EMG, muscle MRI and muscle biopsy) were retrospectively retrieved and analyzed. All patients were examined by at least one of the authors in specialized neuromuscular departments. Diagnostic skeletal muscle biopsies were obtained, processed for standard histological and immunochemical studies and fixed for electron microscopy as previously described [11].
Summarized clinical findings. DMM: delayed motor milestones; F: female; FH: follicular hyperkeratosis; FVC: forced vital capacity;LL: lower limbs; M: male; m: months; N/A: non-applicable; UL: upper limbs; y:years
Informed consent was obtained from all patients in agreement with local ethical committees and with the 1964 Helsinki declaration and its later amendments (NIH, National Institute of Neurological Disorders and Stroke (NINDS), Institutional Review Board (Protocol 12N0095)).
Genetic analysis, bioinformatics analysis and variants interpretation
Details on genetic testing and bioinformatics ana-lysis can be found in Supplemental data. Pathogenicity of variants was determined according to current ACMG guidelines [12]. Variants were filtered out according to their allele frequency (≤1%) as reported in the GnomAD database (http://gnomad.broadinstitute.org/). We then evaluated each variant considering a review of the literature, the location of the variant in the gene and the corresponding protein, the in silico prediction tools (Polyphen2, SIFT, GVGD and CADD for missense variants and SpliceSiteFinder-like, MaxEntScan, NNSPLICE, GeneSplicer and Human Splicing Finder for splicing variants) and functional studies when available. SuSPect method (http://www.sbg.bio.ic.ac.uk/suspect/) was also used for prediction. All variants considered as pathogenic and likely pathogenic have been confirmed by a second independent method (Sanger sequencing).
Collagen VI immunolabelling
Dermal fibroblasts from eight index patients and one control individual were cultured to confluency in DMEM (Gibco) supplemented with 10% FBS (Biosera), penicillin/streptomycin (5700U Pen/5700μg Strep; Gibco), and 50μg/ml L-Ascorbic acid-2-phosphate (Sigma). Fixed cells were immunostained using either the polyclonal antibody Ab6588 (Abcam) as initially described in [13], or an α3(VI) specific antibody (HPA010080; Sigma-Aldrich). For the latter, cells were fixed with cold methanol prior to immunostaining. Confocal imaging was performed on a Nikon Ti2 microscope equipped with a motorized stage and a Yokogawa CSU-W1 spinning disk head coupled with a Prime 95 sCMOS camera (Photometrics). Z-stacks were obtained using a 0.15μm steps, with a 40x/1.30 NA oil-immersion objective. Images were acquired with the same exposure setting, using the Metamorph software (Molecular Devices), and subsequently analyzed using Fiji [14]. Stacks were merged and Z projections (Sum slices) were obtained.
RESULTS
Genetics
Through Next Generation Sequencing we identified the c.7447A>G p.(Lys2483Glu) variant in 16 patients, 5 females and 11 males ranging from age 2 to 77 years (mean 37.9±24.4) at last examination (Table 1).
The COL6A3 c.7447A>G missense variant (rs139260335 in dbSNP) is located in exon 36. At the protein level, the substitution affects the C1/11th Von Willebrand factor A domain (VWA 11), located in a non-helical domain, and leads to an amino acid change with no predicted significant post-translational modifications.
This variant was found either in compound heterozygosity associated with another exonic deletion, truncating or splicing COL6A3 variant (n = 12) or in homozygous state (n = 4) (Table 2). Segregation studies were performed in eight families and confirmed that the tested parents were unaffected heterozygous carriers (Fig. 1).
Genetics and ancillary tests. COL6-RM: Collagen VI related myopathies; FSV: fiber size variation; IN: internalized nuclei; LL: lower limbs; N/A: non-applicable; NCS: nerve conduction studies; UL: upper limbs; WB: Western Blot
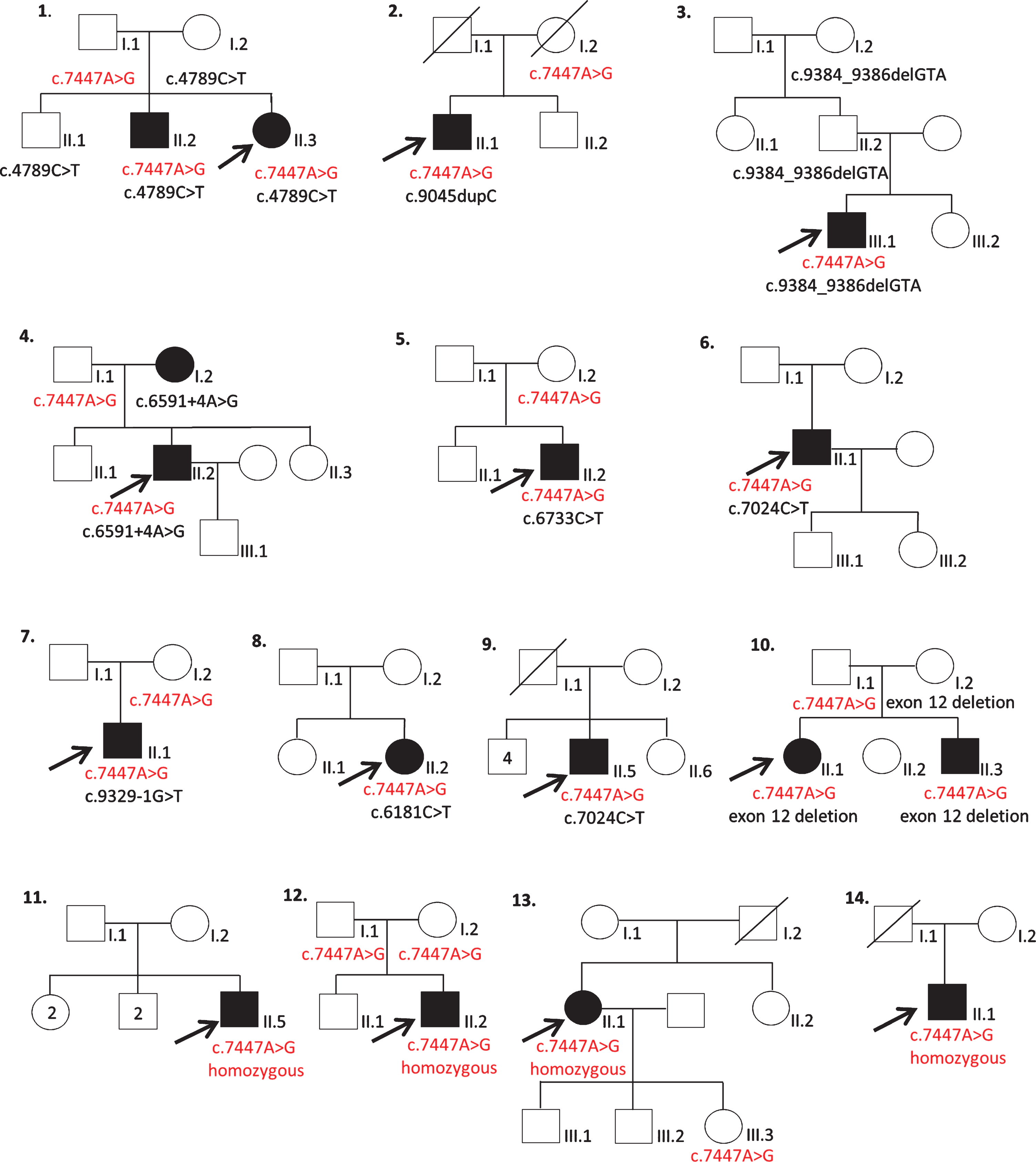
Family pedigrees. Black filled symbols: affected individual. Crossed symbols: deceased individual.
Four patients (11-II.5, 12-II.2, 13-II.1 and 14-II.1) were homozygous for the c.7447A>G COL6A3 variant. Patient 13-II.1’s daughter was a healthy heterozygous carrier. No segregation studies could be performed for patients 11-II.5, 12-II.2 and 14-II.1. Patient 13-II.1 also harbored a heterozygous TNXB variant c.4535_4552del, p.(Asp1512_Val1517del), whose allelic frequency was found to be low in gnomAD (13 allele count/275252) and reported as probably pathogenic in one patient in the LOVD database (reference #0000528445) associated with a known recessive pathogenic TNXB variant. No second TNXB variant was identified in this patient.
Furthermore, no other variants that could affect splicing, were found in the COL6A1-A2-A3 genes after sequencing of the coding regions from cultured skin fibroblasts for patients 12-II.2, 13-II.1 and 14-II.1 harboring the c.7447A>G at the homozygous state (no skin biopsy was available for patient 11-II.5) (data not shown). Whole exome sequencing (WES) was performed in three compound heterozygous patients (7.II.1, 8-II.2, 10-II.1), thereby excluding additional pathogenic variants in other disease-causing genes.
Regarding patient 11-II.5, NGS panel for analysis of genes related to LGMD, distal, myofibrillar panels including NEB and ACTA1 genes (responsible for most forms of nemaline myopathy) disclosed no pathogenic variants.
Clinical presentation of compound heterozygous patients
Ten (83% ) patients had first recognition of symptoms at birth or in early childhood, one around puberty and one in adulthood. Family history was non-contributory. The most commonly recognized presenting symptoms were delayed motor development, waddling gait, poor sports performance and joint contractures. Decreased fetal movements were noted in two patients. Gait acquisition was delayed in two patients (5-II.2 and 10-II.3) who reached independent ambulation at 18 and 21 months, respectively. Two additional patients (8-II.2 and 10-II.1) started walking at 14 months and were noted to have a waddling gait. Muscle weakness was present in all patients with predominant axial and proximal involvement in 11 (92% ), MRC grade 3-4/5. Prominent distal weakness was observed in one patient, afflicting finger flexors and extensors in the upper limbs and ankle dorsiflexion and eversion in the lower limbs (MRC 3/5). Independent ambulation was maintained in all patients although three patients (25% ) required assistance (Table 1).
Joint contractures were present in all patients, mainly involving finger flexors, elbows and ankles. Rigid spine was noted in four patients (33% ). Scoliosis was uniformly absent. Distal hyperlaxity was observed in four patients (33% ). Five patients (42% ) were found to have cutaneous abnormalities including hyperkeratosis, keloid scars and velvety skin.
Forced Vital Capacity was measured in 15 patients and showed mild restrictive respiratory insufficiency in three of them (1-II.1, 3-III.1 and 9-II.5) but they did not need ventilatory support (Table 1). One patient (4-II.2) required a pacemaker due to a left bundle branch block at age 40 years.
Serum Creatine Kinase (CK) levels were mildly to moderately elevated in all patients (range 120–900 IU/L). Electromyography (EMG) was performed in 10 patients, which revealed predominantly myopathic findings in seven patients. Patients 8-II.2 and 10.II.1 had normal nerve conduction studies (NCS). EMG was limited due to poor tolerance in patient 8-II.2. For patient 10.II.1, EMG at age 5 years revealed slightly increased duration and amplitude and mildly reduced recruitment of the left vastus lateralis. Lastly, for patient 9-II.5, NCS showed absent peroneal motor response recorded at the extensor digitorum brevis bilaterally and EMG showed small motor units suggestive of a myopathic pattern in the proximal upper limbs, but neurogenic changes of the distal and proximal legs at age 55 years. Muscle MRI (Fig. 2A and Supplementary Figure 1) was performed in eight patients and showed characteristic COL6-RM findings in seven, namely fatty replacement starting around the fascia surrounding the vastus lateralis (“outside in”) and the so-called “central cloud” pattern in the rectus femoris. Two patients’ MRI showed fatty replacement of pelvic girdle and proximal leg muscles without the characteristic previously mentioned findings. Eight patients underwent a muscle biopsy, revealing prominent dystrophic features in seven (87.5% ).
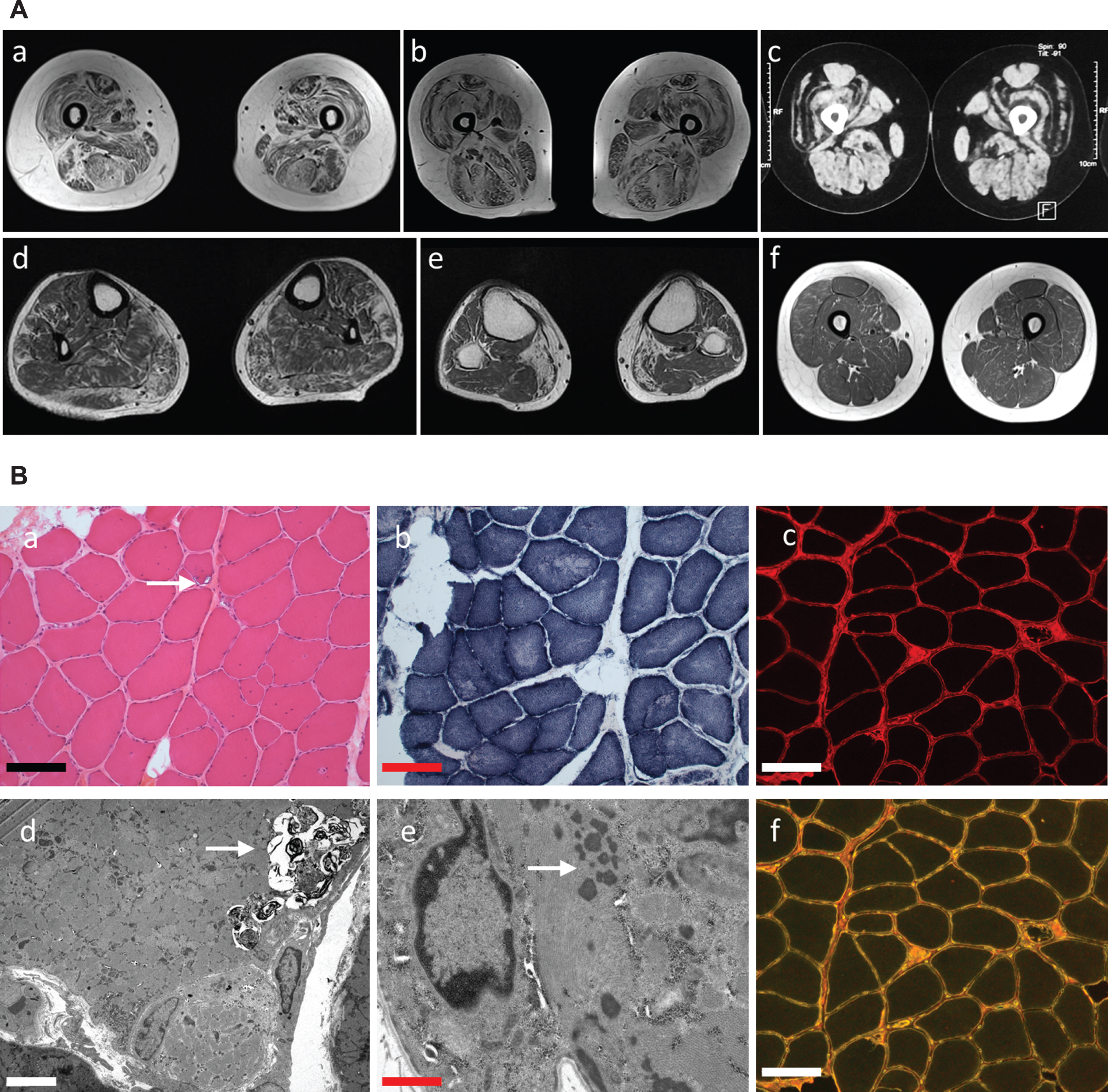
A. Muscle imaging. Axial T1-weighted images. MRI from patients 1-II.1 (a) 6-II.1 (b) and 13-II.1 (c) show typical COL6-RM radiological findings, including fatty replacement starting around the fascia surrounding the muscle and the so-called “central-cloud” affecting the rectus femoris. (d,e) MRI from patient 11-II.5 reveals atrophy and fatty infiltration of distal lower limb muscles mainly affecting medial gastrocnemius and peroneus. (f) MRI from patient 12-II.2 showed no major muscle atrophy and mild fatty replacement of anterior and posterior thigh muscles. B. Muscle biopsy from patient 11-II.5. Hematoxylin and eosin (a) transversal frozen section show mild fiber size variation, numerous internalized nuclei and few rimmed vacuoles (arrow), while NADH-TR (b) reveals diffuse areas lacking oxydative activity. Note slightly irregular or discontinuous COLVI immunostaining (c). (d) Myofibrillar disorganization, autophagic vacuoles (arrow-d) and nemaline rods (arrow-e) are observed by electron microscopy (d,e). (f) COLVI and perlecan co-immunostaining. Scale bars: (a-c,f) 10μm; (d) 15μm; (e) 1μm.
Clinical presentation of homozygous patients
Recognition of first symptoms was in childhood in all four patients. Patient 11-II.5 presented with foot deformities and steppage gait first recognized at age 10 years. Patient 12-II.2 presented with equinovarus feet and toe-walking since early-childhood, patient 13-II.1 presented with abnormal gait since childhood and patient 14-II.1 presented since early childhood with delayed motor milestones (delayed gait acquisition) and difficulty running. The latter three patients were found to have proximal weakness, while patient 11-II.5 had predominant distal weakness and bilateral scapula alata. Patient 14-II.1 had finger flexor and Achilles contractures. Joint contractures were absent in the remaining three patients, aside from mild ankle contractures in patients 12-II.2 and 13-II.1 and none of them had distal hyperlaxity, skin abnormalities or respiratory involvement. Cardiac examination was normal in patients 11-II.5, 13-II.1 and 14-II.1, while echocardiogram from patient 12-II.2 revealed mild aortic insufficiency at age 13 years. All patients maintained ambulation but patient 14-II.1, currently 77 years-old, requires assistance (walker) since age 74.
CK levels were strikingly elevated in patients 11-II.5 and 12-II.2, ranging from 700 to 2000 IU/L, while patients 13-II.1 and 14-II.1 had normal CK levels. Muscle MRI from the first two patients revealed non-typical COL6-RM findings (Fig. 2A). Indeed, fatty infiltration of posterior compartment of distal lower limb muscles was observed in patient 11-II.5 and mild fatty infiltration of proximal limb muscles was noticed in patient 12-II.2. Muscle MRI from patients 13-II.1 and 14-II.1 showed typical COL6-RM findings involving vastus lateralis and rectus femoris.
Muscle biopsy from patient 11-II.5 showed inter-nalized nuclei, myofibrillar disorganization, autoph-agic vacuoles and nemaline rods (Fig. 2B), while dystrophic changes were observed for patients 12-II.2 and 13-II.1 and muscle biopsy from patient 14-II.1 revealed only mild atrophy without necrosis or regeneration and fiber size variation.
Collagen secretion in cultured skin fibroblasts
Collagen VI immunolabelling was performed us-ing two different antibodies, on fixed dermal fibroblasts derived from five compound heterozygous patients (Table 2, Fig. 3 and Supplementary Figure 2), and revealed reduced collagen VI secretion compared to control cells, in which a dense network of deposited collagen VI was detected. Since patients 1-II.2, 2-II.1 and 6-II.1 harbor a second pathogenic variant introducing a premature termination codon that should lead to transcript degradation via the nonsense-mediated decay, the COLVI staining detected most likely reflects the protein synthesis sustained by the missense-bearing allele. Similarly, the second pathogenic variant carried by patients 3-III.1 and 4-II.2 (delins and exon deletion, respectively) should impair the assembly of monomers and/or secreted microfibrils. These results suggest that the missense variant does not significantly prevent COLVI assembly and secretion. Interestingly, COLVI immunolabelling of fibroblasts from three homozygous patients revealed a rather normal secretion pattern (patients 11-II.5 and 13-II.1) or reduced secretion (14-II.1) (Fig. 3 and Supplementary Figure 2). Accordingly, COLVI immunostaining on muscle biopsy from patient 11-II.5 was also rather preserved, with only focal points of discontinuous signal (Fig. 2c and 2f).
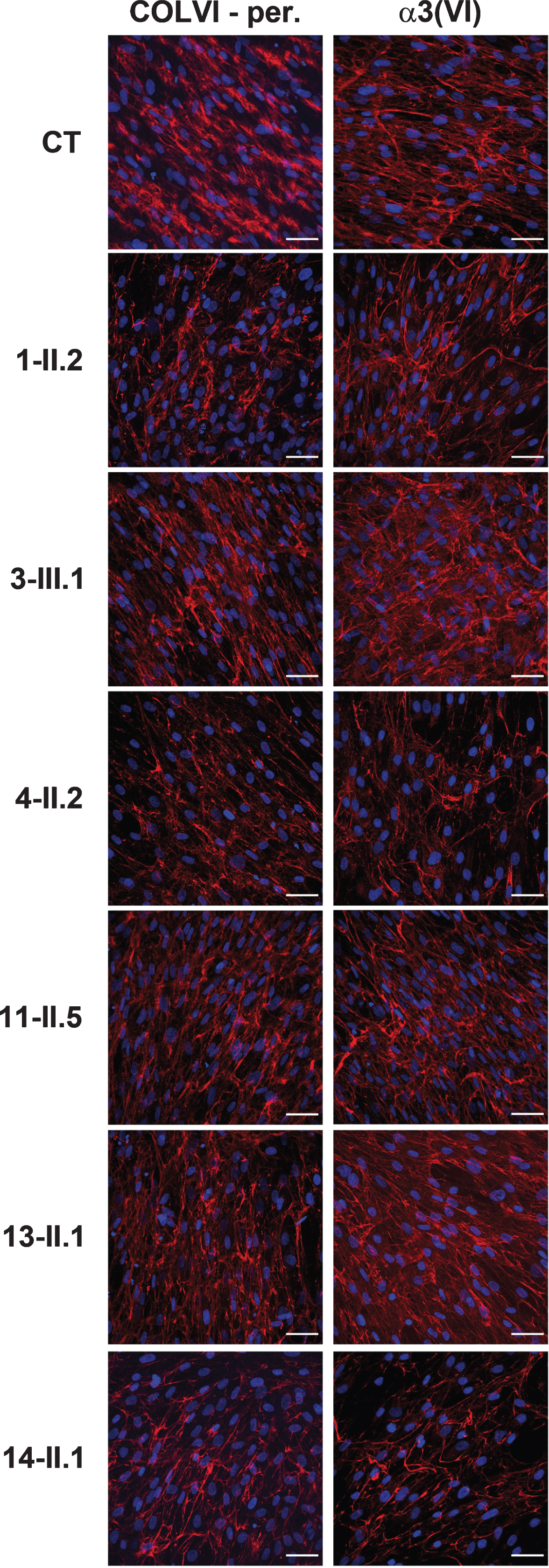
Confocal imaging of COLVI secretion in fixed dermal fib-roblasts from a control individual (CT) and 6 patients. COLVI (red) was detected with two polyclonal antibodies: Ab6588 (Abcam) on non-permeabilized cells (left panel) or an α3(VI) chain specific (Sigma) on methanol-fixed cells (right panel). Nuclei are identified by DAPI staining (blue). In three compound heterozygous patients (1-II.2, 3-III.1 and 4-II.2) reduced collagen VI secretion was observed. Imm-unolabelling of fibroblasts from homozygous patients 11-II.5 and 13-II.1 revealed a rather normal secretion, while it was clearly redu-ced in the culture from homozygous patient 14-II.1. Scale bars = 50μm.
DISCUSSION
COL6-RM diagnosis is based on the combination of clinical presentation (i.e. muscle weakness, prominent contractures, characteristic cutaneous abno-rmalities and variable respiratory involvement), muscular imaging, immunohistochemical examination of collagen VI on muscle biopsy and fibroblasts and analysis of the COL6A1-3 genes.
Although the first pathogenic variants detected in UCMD were recessive null variant, leading to an absence of collagen VI in muscle biopsy sections and in cultured dermal fibroblasts [15], de novo dominant mutations in the COL6A1-3 genes are responsible for a large proportion of UCMD cases [1, 16–18]. In BM, most patients harbor heterozygous dominantly acting pathogenic variants, typically involving glycine residues of the Gly-X-Y motif at the N-terminal end of the triple helical domain that exert a dominant-negative effect on the tetramer structure [6, 19–21]. Nonetheless, recessive mutations in the COL6A1-3 genes have also been detected in patients with typical BM [22, 23], most of them carrying a null variant on one allele in heterozygosity with a missense mutation on the other.
Interpretation of genetic variants can be challenging and many variants in the COL6A1-3 genes have not yet been fully characterized at the functional level. Such is the case of missense variants affecting regions outside of the triple helix. Furthermore, there is a number of patients with a compatible clinical and muscle imaging phenotype who have no detectable pathogenic variants in the three COL6 genes coding sequences [24, 25]. In that sense, 5’/3’ UTR regulatory elements or deep intronic splice mutations can go undetected by standard sequencing approaches, and muscle or fibroblasts RNA sequencing can be useful to detect these pathogenic variants [26].
Along these lines, the interpretation of the COL6A3 c.7447A>G variant is complex. Its allelic frequency is 171/277005 and one homozygous individual is reported in the GnomAD database. It is most prevalent in the non-Finnish European (0.001) and Latino (0.0007) populations. Its high allelic frequency raises questions about its pathogenicity, which remains unclear so far. Its predicted effect on SuSPect method (http://www.sbg.bio.ic.ac.uk/suspect/) [27] points to a low pathogenicity score (8 out of 100). The CADD score for this variant is 22.5 and it is classified as likely pathogenic or variant of unknown signification in LOVD, Clinvar and likely pathogenic in the HGMDPro-database.
We report twelve patients carrying the c.7447A>G COL6A3 variant in compound heterozygosity with a second COL6A3 variant. Most of these patients presented since childhood with proximal weakness associated with joint contractures, variable presence of rigid spine and skin abnormalities. All remained ambulatory at a mean age of 34.7±21.5 and mild respiratory involvement was detected in three. This phenotype would be consistent with Bethlem myopathy (BM) [4, 7]. Muscle MRI showed typical COL6-RM findings in seven out of eight patients and collagen VI deposition in the extracellular matrix was reduced in dermal fibroblasts from five patients. WES analysis in three compound heterozygous patients excluded additional pathogenic variants in other neuromuscular disease-causing genes.
Conversely, homozygous patients exhibit strikin-gly different clinical features. Two out of four did nothave a typical COL6-RM phenotype and presented with foot deformities, wing scapula and distal weakness or abnormal gait with toe-walking without major contractures, distal hyperlaxity, skin abnormalities or respiratory involvement. Moreover, CK levels of these two patients were strikingly high for COL6-RM, muscle MRI disclosed non-specific findings including only distal involvement in one, and muscle biopsy from the one patient showed myofibrillar disorganization, autophagic vacuoles and nemaline rods. Splicing defects were excluded after sequencing of the COL6A1-A2-A3 coding regions in three patients homozygous for the c.7447G>A variant.
The COL6A3 c.7447A>G variant has been previously reported in compound heterozygous patients [5, 28–31] with a clinical spectrum ranging from a mild phenotype when associated with a missense variant [30], to an intermediate phenotype with childhood onset and respiratory involvement [5] and a severe Ullrich-like phenotype [29], when associated with a second truncating COL6A3 variant. To our knowledge, six homozygous patients have been reported so far [5, 32]. Panadés-de Oliveira et al. [5] reported two homozygous siblings. The index case had proximal weakness, elevated CK levels (1000 IU/L) and dystrophic findings and rimmed vacuoles on muscle biopsy. The patient’s sibling had asymptomatic significantly increased CK levels (4000 IU/L) detected at mid-age. Both had typical COL6-RM findings on muscle MRI findings. This study reported one additional homozygous patient who also carried a COL6A1 c.2435-2A>G pathogenic splicing variant [5]. Interestingly, rimmed vacuoles were also found on the muscle biopsy from our patient 11-II.5.
The patient reported by Hunter et al. [29] presented with club feet, abnormal gait and lipoatrophy, developed scoliosis and hamstrings, ankles and feet contractures with overlapping toes. EMG studies revealed a chronic motor neuropathy and muscle biopsy disclosed abnormal myofibrillar architecture with fiber type grouping. Recently, Stavusis et al. [32] reported two siblings with mild proximal weakness and joint contractures carrying three COL6A3 variants: the c.7447A>G variant in homozygosity combined with the heterozygous frameshift variant c.8074delT, p.(Tyr2692MetfsTer15). Strikingly, they also reported a third patient, carrying the COL6A3 c.7447A>G variant in homozygosity, with proximal and distal weakness, diminished ankle reflexes and axonal polyneuropathy, but no further biological or genetic analysis regarding this polyneuropathy is reported. Interestingly, collagen VI is expressed in peripheral nerve [33–35]. Nonetheless, COL6A1-A2-A3 pathogenic variants have not been found associated with hereditary neuropathies. On another note, muscle MRI of this patient showed diffuse and severe fatty infiltration without typical COL6-RM findings and CK levels were also elevated from two to four times the normal values. Unfortunately, no collagen VI immunolabeling on muscle biopsy or analysis of collagen VI production in dermal fibroblast cultures are reported for any of these homozygous patients.
Stavusis et al. [32] have also reviewed all the rep-orted patients to date, and conclude that for compound heterozygous patients, the phenotype severity entirely depends on the second pathogenic variant. The authors speculate that this variant could perturb the binding properties of the protein and thus the stability of the heterotrimer and modulate the phenotype when present along with other pathogenic variants, as is the case forthe patients with the c.8074delT variant, which leadsto a premature termination of the protein. Nonetheless, the occurrence of another deep intronic pathogenic variant in the COL6A1-3 genes cannot be excluded [32]. Strikingly, reported homozygous patients exhibit different clinical phenotypes such as peripheral neuropathy [29, 32], asymptomatic hyperCK [5] or limb-girdle myopathy with joint contractures [5, 32], associated in some cases with a non-typical COL6-RM histopathological presentation including rimmed vacuoles [32] or abnormal myofibrillar architecture with fiber type grouping [29].
At the functional level, the determinant role of the α3(VI) chain in the monomer assembly is well established [36]. However, the exact contribution of the VWA C1 domain, harboring the mutated Lysine residue at position 2483, remains poorly understood, although it has been shown to be important for chain recognition and assembly of triple helical molecules [37]. Immunostaining of muscle biopsy and fibroblast cultures show that the missense variant does not prevent COLVI deposition in the ECM, although further analyses, such as Western blotting, should be performed to obtain quantitative data. VWA domains being protein-protein interaction modules, the missense variant may alter interaction with binding partners as suggested by Stavusis et al. [32] but further investigations are needed to support this hypothesis experimentally.
In conclusion, this work expands the clinical and molecular spectrum of COLVI-related myopathies and is focused on the complex interpretation of the frequent COL6A3 c.7447A>G variant and the associated clinical findings. Its high allele frequency and the different phenotypes observed in the homozygous patients reported so far, raise questions about the pathogenicity of this variant in homozygosity. Our results suggest that the COL6A3 variant c.7447A>G may act as a modulator of the clinical phenotype. An in-depth analysis of clinical features and ancillary tests is mandatory in order to interpret the genetic analysis. For homozygous patients with a compatible COL6-RM phenotype, including clinical, MRI findings or altered collagen VI secretion, we recommend to thoroughly search for an additional genetic variant in any of the COL6A1-3 genes, as in the cases reported by Panades et al. [5] and Stavusis et al. [32], including deep intronic variants leading to aberrant splicing that may go undetected in NGS based panels [26]. Thus, analysis of mRNA transcripts could be useful. In the event of a non-compatible phenotype such as atypical clinical findings, strikingly elevated CK levels, non-pathognomonic COL6-RM muscle MRI findings, further genetic analysis would be advisable to exclude alternative causes such as inherited neuropathies or other forms of myopathies. Further analysis of homozygous patients with detailed clinical and ancillary tests data together with molecular and genetic studies would help to elucidate whether this variant is disease-causing or not. This would be of major importance to allow precise diagnosis, accurate management and genetic counseling for the affected patients and families.
Footnotes
ACKNOWLEDGMENTS
We thank the patients and their families for participating in our research study and Christopher Mendoza, Christine Jones, and Gilberto (“Mike”) Averion for their help in the clinical setting. The monoclonal antibody HFN7.1 developed by The University of Texas Health Science Center at San Antonio was obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242. We also like to thank Dr. Guillaume Lamotte and Dr. Craig M. Zaidman for their clinical support. We thank Généthon (Evry, France) for deriving fibroblast cultures from skin biopsies.
FUNDING
The work in C.G. Bönnemann’s laboratory is supported by intramural funds from the NIH National Institute of Neurological Disorders and Stroke. V. Allamand’s group is supported by intramural funds from the Association Institut de Myologie, and Inserm.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.