
Abstract
Myotonic dystrophy type 1 (DM1) is the most common monogenetic muscular disorder of adulthood. This multisystemic disease is caused by CTG repeat expansion in the 3′-untranslated region of the DM1 protein kinase gene called DMPK. DMPK encodes a myosin kinase expressed in skeletal muscle cells and other cellular populations such as smooth muscle cells, neurons and fibroblasts. The resultant expanded (CUG)n RNA transcripts sequester RNA binding factors leading to ubiquitous and persistent splicing deregulation. The accumulation of mutant CUG repeats is linked to increased activity of glycogen synthase kinase 3 beta (GSK3β), a highly conserved and ubiquitous serine/threonine kinase with functions in pathways regulating inflammation, metabolism, oncogenesis, neurogenesis and myogenesis. As GSK3β-inhibition ameliorates defects in myogenesis, muscle strength and myotonia in a DM1 mouse model, this kinase represents a key player of DM1 pathobiochemistry and constitutes a promising therapeutic target. To better characterise DM1 patients, and monitor treatment responses, we aimed to define a set of robust disease and severity markers linked to GSK3βby unbiased proteomic profiling utilizing fibroblasts derived from DM1 patients with low (80– 150) and high (2600– 3600) CTG-repeats. Apart from GSK3β increase, we identified dysregulation of nine proteins (CAPN1, CTNNB1, CTPS1, DNMT1, HDAC2, HNRNPH3, MAP2K2, NR3C1, VDAC2) modulated by GSK3β. In silico-based expression studies confirmed expression in neuronal and skeletal muscle cells and revealed a relatively elevated abundance in fibroblasts. The potential impact of each marker in the myopathology of DM1 is discussed based on respective function to inform potential uses as severity markers or for monitoring GSK3β inhibitor treatment responses.
INTRODUCTION
Myotonic dystrophy type 1 (DM1; MIM: 160900) is the most common monogenetic muscular disorder of adulthood with an estimated worldwide prevale-nce of 5/100,000, ranging from 0.5 in Taiwan up to 18.1/100,000 in Croatia [1]. In addition, DM1is highly prevalent in Canada (Saguenay-Lac-Saint-Jean) where its carrier rate reaches 1/550 [2, 3]. The disease is characterised by progressive muscle atrophy and weakness, often combined with myotonia, fatigue, respiratory insufficiency, and speech and swallowing difficulties [4, 5]. DM1 represents a mul-tisystemic disorder and affects multiple organs inc-luding the central nervous system, heart, eye, skin, gastrointestinal and reproductive tracts, as well as the endocrine and immune systems [5]. Traditionally, DM1 is categorized according to phenotype and age of onset. However, this classification into congenital, juvenile, childhood, adult and late onset is complicated by significant intrafamilial and interfamilial variability [5, 6].
DM1 is an autosomal dominant disorder, caused by a CTG repeat expansion [(CTG)n] in the 3′-untranslated region of the DM1 protein kinase gene (DMPK). DMPK is located at position 19q13.32 and encodes a myosin kinase expressed in skeletal muscle [7]. Expanded alleles display microsatellite instabi-lity towards expansion in germline cells (causing genetic anticipation) and somatic cells (contributing to disease progression and phenotype variability [8–11]). The resultant expanded (CUG)n RNA transc-ripts cause the DM1 phenotype by sequestering RNA binding factors and leading to ubiquitous and persistent splicing deregulation [5, 12– 17]. This toxic (CUG)n RNA effect is mediated by gain of function of combined CUGBPElav-like family member 1 (CELF1) and loss of function of muscleblind like splicing regulator (MBNL) due to clustering of both proteins [15–17]. Several aberrant splicing events have been linked to muscular [15, 18– 21], cardiac [15, 23], neurocognitive [24, 25] and endocrine [15, 26– 28] features of DM1.
Glycogen synthase kinase 3 beta (GSK3β) activity is significantly increased in the skeletal muscles of DM1 patients [29]. This increase in GSK3β activity is linked to the accumulation of mutant CUG repeats and, by promoting the phosphorylation of cyclin D3 at T283, leading to degradation and resultant in muscle weakness. GSK3β is a highly conserved and ubiquitous serine/threonine kinase with an important pleiotropic role in pathways regulating inflammation, metabolism, oncogenesis, neurogenesis and myogenesis [29–34]. Pharmacological inhibition of GSK3β ameliorates defects in myogenesis, muscle strength and myotonia in DM1 mice (HSALR) [29] further supporting the emergence of GSK3β as a novel and promising therapeutic target for DM1 [35]. To better characterise DM1 patients, and monitor treatment responses, it will be necessary to define a set of robust disease and severity markers.
PATIENTS, MATERIAL AND METHODS
In this study, we aimed to identify DM1 protein markers reflecting activation of pathways, such as GSK3β overactivation, in patient-derived fibroblasts, a suitable cell model to study the molecular etiology of neuromuscular diseases [36]. Proteomic profiling was performed on fibroblasts derived from DM1-patients with a mild/late onset [(CTG) 80– 150] and a severe/congenital [(CTG) 2600– 3600] DM1 phenotype. Obtained data were filtered for dysregulated proteins that are known to be modulated by GSK3β or involved in GSK3β-dependent processes.
Ethical considerations
This study has been ethically approved as a sub-study as part of OPTIMISTIC (reference: NRES Committee North East - Sunderland 13.NE.0342) and PHENODM1 (reference: NRES Committee North East – Tyne & Wear South 15.NE.0178). Patients were consented and recruited at Newcastle upon Tyne NHS Foundation Trust and all procedures leading to these results complied with the Good Clinical Practices and the Declaration of Helsinki.For skin biopsies studies, informed consent was obtained from all patients.
Fibroblasts and cell culture
Fibroblasts were isolated from fresh donor skin biopsies following standardised EuroBioBank protocols (www.eurobiobank.org/biobanking-sops): biopsies were washed with sterile phosphate-buffered saline (PBS) and digested at 37°C for 15 minutes with 2.5% trypsin (Thermo Fisher Scientific) and a further 90 minutes with 0.5% collagenase (Type IV, Sigma-Aldrich). Fibroblasts were proliferated in Ham’s F-10-Complete Medium (Thermo Fisher Sci-entific) supplemented with 20% foetal bovine serum (FBS, SeraLab – Bioreclamation IVT), 2% pen-icillin-streptomycin (Thermo Fisher Scientific), 1% GlutaMAX™ (Thermo Fisher Scientific) and 1% fungizone (Thermo Fisher Scientific). Once fibroblast cells attained sufficient confluency, they were frozen and stored long-term in liquid nitrogen. For further experiments including proteomic profiling, cells were cultured as described above until a confluence of 70%, harvested by scraping from culture flasks, washed twice with ice-cold PBS and cell pellets were snap-frozen in liquid nitrogen and stored at – 80°C until further processing.
In total 12 fibroblast samples were included in our proteomic study: 3 DM1 fibroblast lines from patients with early onset, severe disease and CTG-repeat expansions of more than 2000 (patients 4– 6:2431, 2683 and 3187 CTG-repeats, respectively), 3 DM1 fibroblast lines from patients with late onset, mild disease and CTG-repeat expansions between 80 and 150 CTG-repeats (patients 1– 3:84, 94 and 140 CTG-repeats, respectively), and a total of 6 sex- and age-matched controls (three controls per patient-group). DM1-patient derived cell lines were obtained from the “MRC Centre Neuromuscular Biobank” (Newcastle upon Tyne, UK) [37]. All patients were adults when the skin biopsies were taken. Clinical data for the individual patients are listed in Table 1.
Clinical findings for the six DM1 patients who provided fibroblasts for the proteomic profiling study. MIRS: Muscular impairment rating scale. Repeat expansions refer to DNA extracted from fibroblasts
Unbiased label-free LC-MS/MS and data analysis
The following reagents were used for LC-MS/MS: Ammonium hydrogen carbonate (NH4HCO3), anhydrous magnesium chloride (MgCl2), guanidine hyd-rochloride (GuHCl), iodoacetamide (IAA), and urea (Sigma-Aldrich, Steinheim, Germany), tris base (Ap-plichemBiochemica, Darmstadt, Germany), sodium dodecyl sulfate (SDS) (Carl Roth, Karlsruhe, Germany), dithiothreitol (DTT), EDTA-free protease in-hibitor (Complete Mini) tablets (Roche Diagnostics, Mannheim, Germany), NaCl, CaCl2 (Merck, Darmstadt, Germany), sequencing grade modified trypsin (Promega, Madison, WI USA), Benzonase® Nuclease (Novagen), a bicinchoninic acid assay (BCA) kit (Thermo Fisher Scientific, Dreieich, Germany). All chemicals for ultra-pure HPLC solvents such as formic acid (FA), trifluoroacetic acid (TFA) and acetonitrile (ACN) were obtained from Biosolve, Valkenswaard, The Netherlands.
Cell lysis, sample clean-up and proteolysis
Cell pellets were lysed in 100μL of lysis buffer (50mM Tris-HCl (pH 7.8), 150mM NaCl, 1 % SDS, and Complete Mini) using a manual glass grinder. Extracts were centrifuged at 6000g for 5min at 4°C to separate cell debris from the protein lysate and protein concentration was determined by BCA assay (according to the manufacturer’s protocol). Cysteines were reduced via the addition of 10mM DTT followed by an immediate incubation at 56°C for 30min. Alkylation of free thiol groups with 30mM IAA was performed at room temperature (RT) in the dark for 30min.
Sample preparation was performed using filter-aided sample preparation (FASP) [38, 39] with some minor changes: 100μg of protein lysate was diluted 10-fold with freshly prepared 8M urea/100mM Tris-HCl (pH 8.5) buffer and placed on a PALL microsep centrifugal device (30KDa cut off) and centrifuged at 13,500g at RT for 20min (all the following centrifugation steps were performed under the same conditions). Three washing steps were carried out with 100μL of 8M urea/100mM Tris-HCl (pH 8.5). For buffer exchange, the device was washed three times with 100μL of 50mM NH4HCO3 (pH 7.8). The digestion buffer (final volume of 100μL) composed of trypsin (1:25w/w, protease to substrate), 0.2M GuHCl and 2mM CaCl2 in 50mM NH4HCO3 (pH 7.8), was added to the concentrated proteins and samples incubated at 37°C for 14h. The resulting tryptic peptides were recovered by centrifugation with 50μL of 50mM NH4HCO3 followed by 50μL of ultra-pure water. Peptides were acidified (pH<3 by addition of 10 % TFA (v/v)). All digests were quality controlled as described previously [40].
LC-MS/MS analysis
1μg of each sample was analyzed using an Ultimate 3000 nano RSLC system coupled to an Orbitrap Fusion Lumos mass spectrometer (both Thermo Sci-entific) in a randomized order to minimize systema-tic errors. Peptides were preconcentrated on a 100μm×2cm C18 trapping column for 10min using 0.1 % TFA (v/v) at a flow rate of 20μL/min followed by separation on a 75μm×50cm C18 main column (both Pepmap, Thermo Scientific) with a 95min LC gradient ranging from 3– 35 % of 84 % ACN, 0.1 % FA (v/v) at a flow rate of 250nL/min. MS survey scans were acquired in the Orbitrap from 300 to 1500 m/z at a resolution of 120,000 using the polysiloxane ion at m/z 445.12002 as lock mass [41], an automatic gain control target value of 2.0×105 and maximum injection times of 50ms. TopSmost intense signals were selected for fragmentation by higher-energy collisional dissociation with an energy of 30 % and MS/MS spectra were acquired in the Iontrap using an automatic gain control target value of 2.0×103 ions, a maximum injection time of 300ms, a dynamic exclusion of 15s and an isolation window of 1.2 (m/z).
Label free data analysis
Data analysis of the acquired label free MS data was performed using the Progenesis LC-MS software from Nonlinear Dynamics (Newcastle upon Tyne, U.K.). Raw MS data was aligned by Progenesis, wh-ich automatically selected one of the LC-MS files as reference. After automatic peak picking, only features within retention time and m/z windows from 0– 95min and 300– 1500m/z, with charge states +2, +3, and +4 were considered for peptide statistics and analysis of variance (ANOVA) and MS/MS spectra were exported as peak lists. Peak lists were searched against a concatenated target/decoy version of the human Uniprot database (downloaded on 22.07.2015 containing 20273 target sequences) using Mascot 2.4 (Matrix Science, Boston, MA, USA), MS-GF+ (beta,v10282), X!Tandem (X!Tandem Vengeance, 2015.12.15.2) and MyriMatch (2.2.140) with the help of searchGUI 3.2.5 [42]. Trypsin was selected as the enzyme, with a maximum of two missed cleavages, carbamidomethylation of cysteine was set as fixed and oxidation of methionine was selected as variable modification. MS and MS/MS tolerances were set to 10 p.p.m and 0.5 Da, respectively.
To obtain a peptide-spectrum match and to max-imize the number of identified peptides and proteins at a given quality, we used PeptideShaker softw-are 1.4.0 (http://code.google.com/p/peptide-shaker/) [43]. Combined search results were filtered at a false discovery rate of 1% on the peptide and protein level and exported using the PeptideShaker features that allow direct re-import of the quality-controlled data into Progenesis. Peptide sequences containing oxidized methionine were excluded from further ana-lysis. Only proteins quantified with unique peptides were exported. For each protein, average of the nor-malized abundances obtained from Progenesis was calculated to determine the ratios between the pat-ient and control cells. Only proteins which were (i) commonly quantified in all the replicates with (ii) a minimum of one unique peptide, (iii) an ANOVA p-value of ≤0.05 (Progenesis) and (iv) an average ratio log2 ≤5.8 or ≥– 0.73 for the low repeats (com-parison 1) and an average ratio log2 ≤5.6 or ≥– 2.3 for the high repeats (comparison 2) were considered as significantly regulated.
“STRING” (Search Tool for the Retrieval of Interacting Genes/Proteins; www.string-db.org) enables the delineation of direct and functionally related protein-protein interactions. Thus, to identify functional interdependences of proteins with altered abundances in diseased cells and tissues, we applied this in silico tool to decipher proteins interacting with the ten marker proteins identified by our proteomic profiling of DM1-patient derived skin fibroblasts.
The Genotype-Tissue Expression (GTEx; www.gtexportal.org) project, representing a comprehensive public resource to study tissue-specific gene expression, was used to address expression of GSK3β and identified markers in DM1 vulnerable tissues including different brain areas, peripheral nervous system, skeletal and cardiac muscle and skin/cultured fibroblasts. The data used for the analyses described in this manuscript were obtained from the GTEx Portal on 20/05.2020 dbGaP accession number phs000424.vN.pN on 12/10/2012.
Immunohistochemistry on murine muscles
Immunological staining of CAPN1 was performed utilizing cardiac and tibialis anterior muscle derived from DMSXL mice (a suitable mouse model of DM1) [44] and wildtype littermates aged two and four months (two animals per group) as described previously [45]. For that purpose, a primary anti-Calpain 1 polyclonal antibody (Invitrogen: #PA5-86056) was used.
RESULTS
Expression of GSK3β in fibroblasts derived from patients with DM1
Proteomic profiles of fibroblasts derived from DM1 patients (early and adult disease onset; Table 1) and respective controls were generated. First, we analysed the proteomic signature for GSK3β within the whole protein extracts obtained from DM1-pa-tient derived fibroblasts. This analysis revealed an increase in GSK3β expression in fibroblasts derived from DM1-patients of both groups; and this effect was more pronounced in cells derived from patients mildly affected by DM1 showing a 2.80-fold increase compared to a 1.62-fold increase in fibroblasts derived from severely affected DM1-patients (severe vs mild=0.58, p-ANOVA=0.110, statistically not significant; Table 2 &3; Fig.1A).
Dysregulation of GSK3β and associated proteins in DM1-patient derived fibroblasts
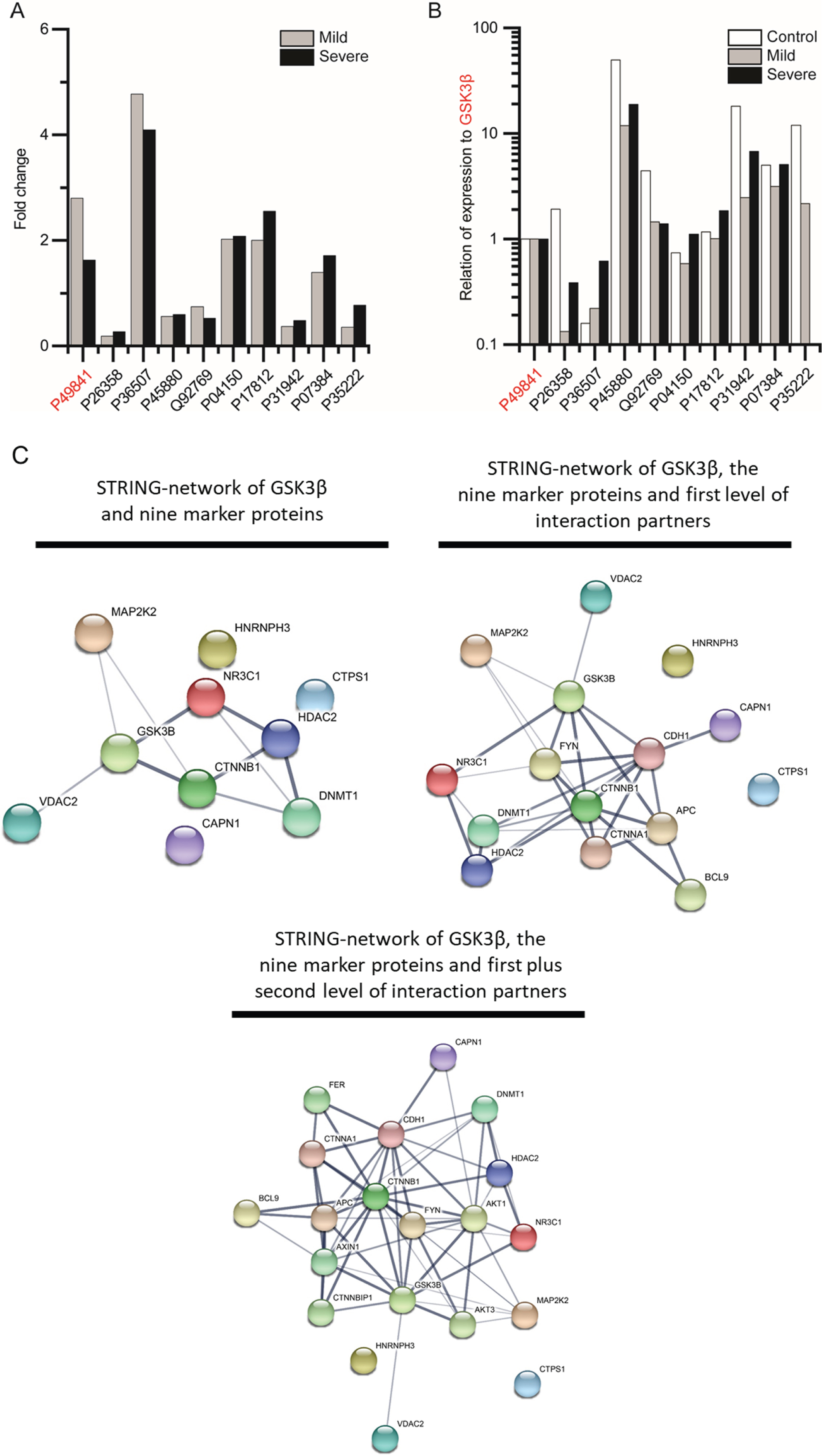
Dysregulation of GSK3β and related proteins identified by unbiased proteomic profiling of DM1-patient derived skin fibroblasts (mildly and severely affected cases). (A) Altered relative expression of GSK3β (P49841; highlighted in red) and nine proteins known to be modulated by its function, thus serving as GSK3β-dependent cellular markers of DM1. (B) Changes in abundance of proteins expressed as a ratio with GSK3β expression in fibroblasts derived from mild and severely affected DM1 patients compared to controls. (C) STRING-based protein interaction networks reflecting interaction network of the nine markers identified, including first and second level binding partners. Thickness of the lines between the nodes refers to the confidence of interaction.
Identification of dysregulated proteins related to GSK3β
Next, we screened these profiles for detection of one of the ∼100 proteins known to be modulated by GSK3β or involved in GSK3β -dependent processes, allowing the identification of 25 dysregulated proteins [31, 30]: 9 of these dysregulated proteins were quantified with a statistically significant p-ANOVA (≤0.05) in both patient groups (Table 2 & Fig.1A) and 16 with a statistically significant p-ANOVA (≤0.05) in only one of the defined groups (Supplementary Table1). Out of the nine proteins, four are decreased (DNMT1, VDAC2, HNRNPH3 & CTNNB1) and five (MAP2K2, HDAC2, NR3C1, CTPS1 & CAPN1) are increased in both patient groups as compared to controls (Table 2 & Fig.1A). To further define a disease marker as a marker of severity, the statistical significance of the change in fold-regulation for each protein between mild and severe patient groups was taken into account. This resulted in the definition of three severity markers (Table 2): HDAC2 is 2.42-fold upregulated in cells derived from mild patients, and 4.86-fold increased in severe patients (p-ANOVA 0.005). Similarly, CTPS1 is 1.99-fold increased in mild patient fibroblasts and 2.55-fold in cells of the severe patient group (p-ANOVA 0.04). Moreover, CAPN1 is 1.39-fold upregulated in cells derived from mild patients, and 1.71-fold increased in severe patients (p-ANOVA 0.002).
Change in expression of dysregulated proteins in relation to GSK3β
To investigate potential stoichiometric changes between GSK3β and the nine substrates dysregulated in DM1-patient derived fibroblasts, we compared the abundance of each protein with GSK3β expression. This comparison revealed decreased ratios for DNMT1, MAP2K2, VDAC2, HDAC2, HNRNPH3 and CTNNB1 in both patient groups compared to controls. For NR3C1 and CTPS1 decreased ratios were observed in the mild group and increased rat-ios in the severe DM1-patient group. For CAPN1, decreased ratios were identified by comparing control and mild patient group, but ratios remained almost unchanged between the controls and the group of severely affected DM1 patients. All protein intensities and calculated ratios to GSK3β are shown in Fig.1B and listed in Table 3.
Abundance of each GSK3β-related protein identified in the proteomics of DM1 patient-derived fibroblasts, as compared to expression levels of GS3Kβ
Protein network analysis
“STRING” protein-network analyses revealed a predicted functional interaction of GSK3β, MAP2K2, CTNNB1, HDAC2, NR3C1, DNMT1& VD-AC2 (Fig.1C). Expanding the interaction network to include first level interactors of our dysregulated proteins revealed a further predicted interplay of 13 proteins, with the addition of CDH1, FYN, CTNNA1, APC and BCL9 as well as connecting CAPN1 to the functional network (Fig.1C). Considering second level binding partners, a functional interplay of 18 proteins was delineated, with the addition of AXIN1, FER, AKT1, AKT3 and CTNNBIP1 (Fig.1C).
In silico tissue expression analysis of dysregulated proteins
To address tissue expression of the identified dysregulated proteins, the GTEx database was used, with a focus on DM1-associated tissues, such as different areas of the central nervous system (amygdala, anterior cingulate cortex, frontal cortex, basal ganglia, cerebellum and cerebellar hemisphere, hippocampus, hypothalamus, substantia nigra and spinal cord), tibial nerve, skeletal and cardiac muscle as well as skin and cultured fibroblasts. Of note, among the different brain areas, all proteins show highest abundance in the cerebellum, including the cerebellar hemisphere. For CAPN1 and MAP2K2 increased abundance was also observed in cortex (Fig. 2). In addition, the nine markers and GSK3β are highly expressed in the tibial nerve, skin and cultured fibroblasts. Compared to the other tissues addressed, moderate abundance in skeletal muscle was observed for GSK3β, VDAC2, CAPN1, CTPS1, NR3C1 and MAP2K2 (Fig. 2).
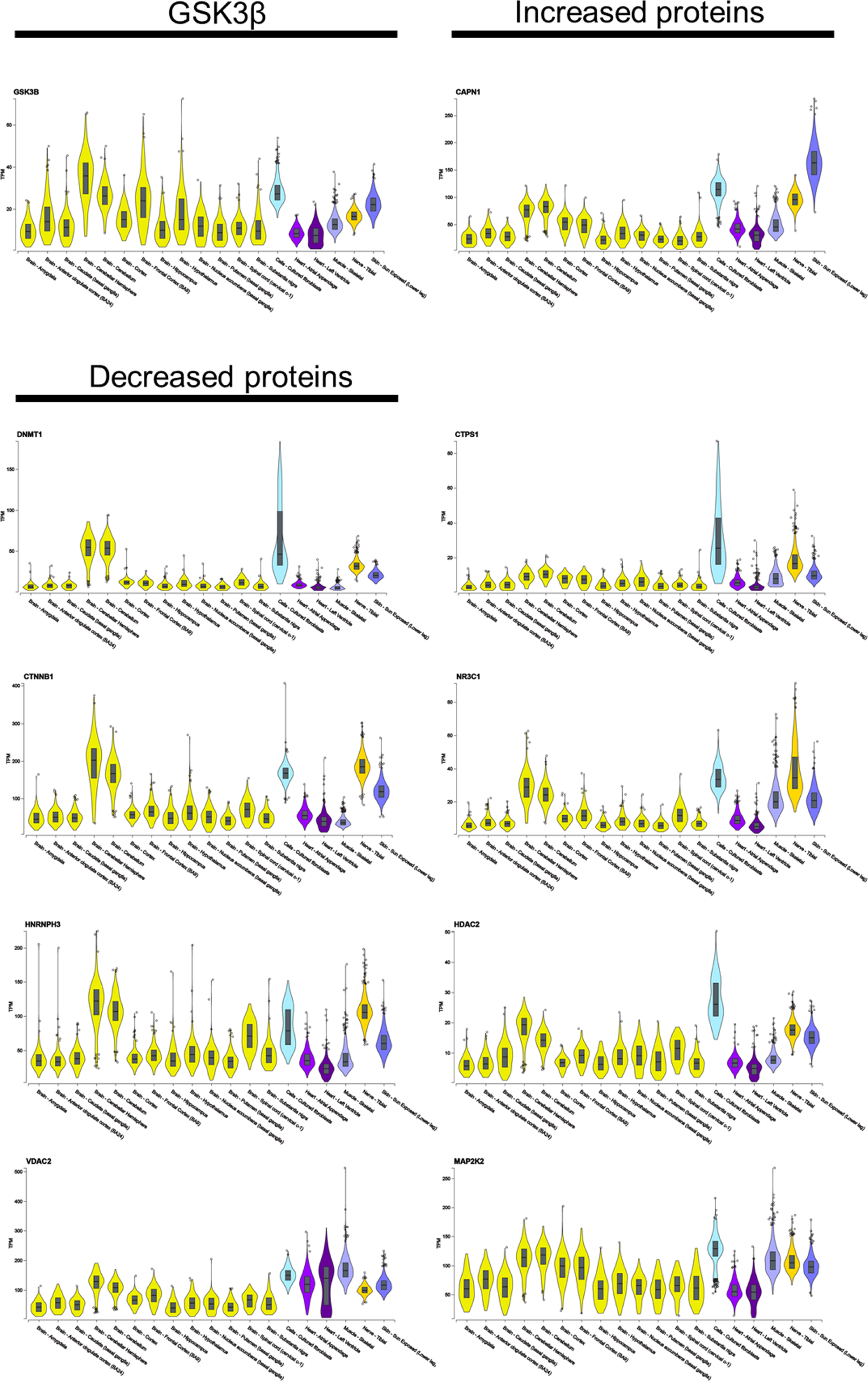
GTEx-based in silico analysis of tissue expression of GSK3β and the nine associated proteins dysregulated in DM1-patient derived skin fibroblasts.
Calpain-1 expression analysis in murine muscle
To verify our proteomic findings, expression CA-PN1 as a paradigmatic protein was studied in cardiac and tibialis anterior muscle derived from DMSXL mice and wildtype littermates (two and four months of age). For both tissues elevated sarcoplasmic CAPN1 level were observed at two and four months of age, respectively (Fig. 3).
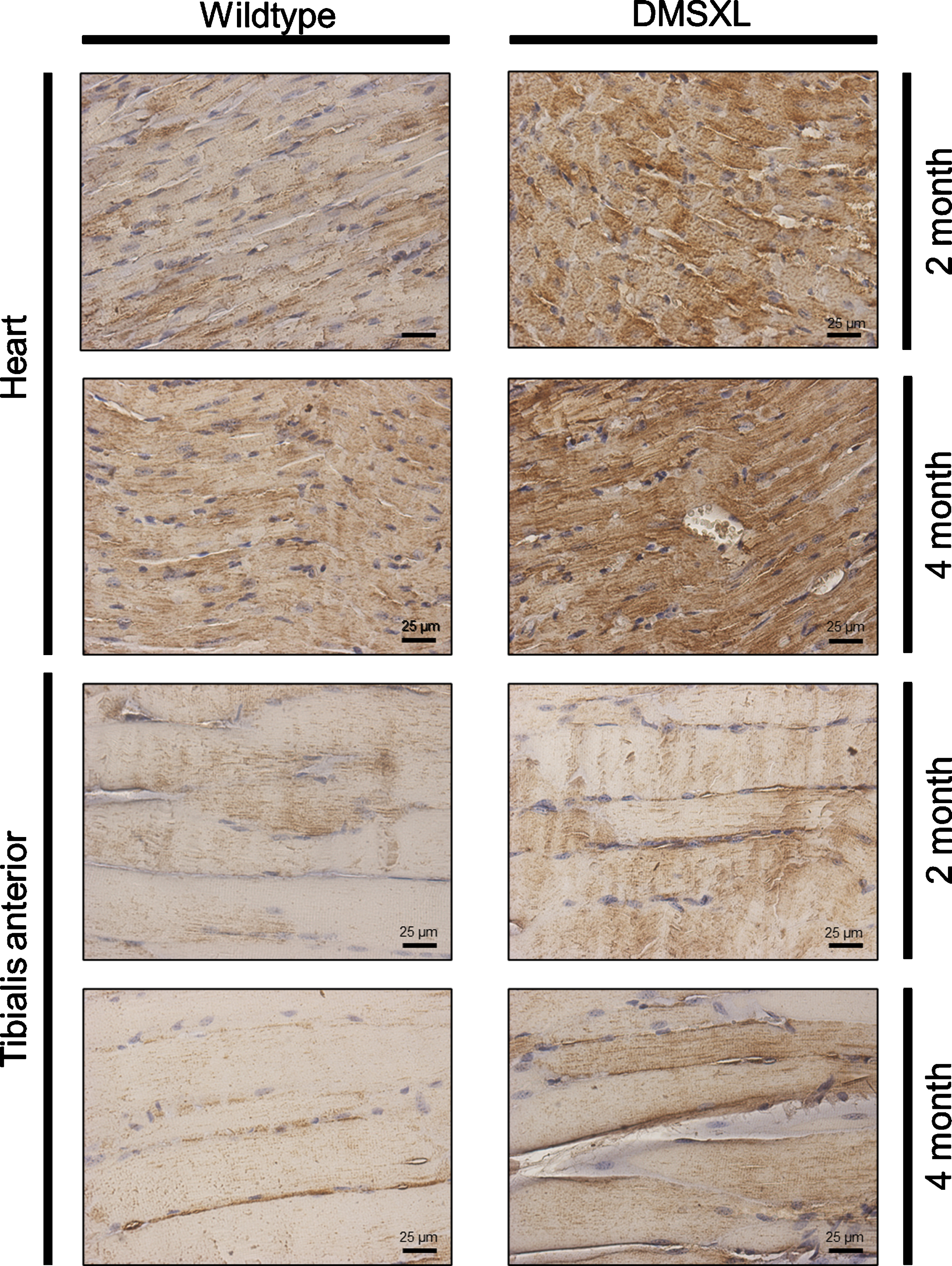
Immunohistochemistry-based analysis of CAPN1 expression in cardiac and tibialis anterior muscle derived from DMSXL mice. Increased immunoreactivity of CAPN1 is detected in cardiac and tibialis anterior muscle (longitudinal sections) of diseased animals compared to controls at the age of two and four months.
DISCUSSION
Proteomic profiling is a powerful tool for the unbiased identification of proteins contributing to neuromuscular disease pathology in cells and tissues derived from patients [46, 47]. Fibroblasts are known to be affected in DM1 [48], therefore, we performed LC-MS/MS-based analysis of the protein signature of DM1 patient-derived fibroblasts to identify proteins that may be markers of DM1 presence or severity.
Nearly 100 proteins are proposed to be substrates for GSK3β, highlighting the role of this kinase as a fundamental regulator of many cellular processes [31–49]. Given that GSK3β-overactivation is found in DM1-patient cells and tissues [29] and that inhibition of this overactivation represents a promising treatment strategy in DM1, we analysed GSK3β abundances in DM1-patient derived cells. This revealed an increase in GSK3β in both mild and severely affected patients. This effect was more pronounced in cells derived from mildly affected DM1-patients, thus in fibroblasts GSK3β protein level does not correlate with CTG-repeat expansion (and the resultant disease-severity classification). However, given that GSK3β-overactivation is modulated by post-translational modification, studies of GS3Kβ-phosphorylation are needed to draw a conclusion regarding a correlation of GSK3β-activation and CTG-repeat expansion/disease severity in DM1-fibroblasts.
After filtering out proteomic data for proteins modulated by GSK3β, we were left with nine proteins that may be potential markers of DM1 disease or severity. To the best of our knowledge, none of the nine proteins has been described as a DM1 blood biomarker thus far. In silico-based interaction studies suggested a down-stream impact on pathways perturbed in DM1, such as AKT-signalling (AKT1 & AKT3) [50]. The known ubiquitous expression of the nine cellular markers hints toward a potential overall involvement in DM1-pathophysiology and moreover suggests that further functional studies or pre-clinical intervention concepts can be performed in DM1-patient derived skin fibroblasts. This postulate is further supported by the proven high expression in human cultured skin fibroblasts as exemplified by or in silico-based expression studies. A comparison of their relative abundance with GSK3β levels showed many proteins had expression that varied with severity. This finding suggests a pathophysiological shift of GSK3β, and some of its substrates, that may allow their use for tracking progression of disease or response to GSK3β inhibitor therapy. The relevance of each GSK3β-related dysregulated protein to DM1 pathophysiology is discussed in subsequent sections:
DNA (cytosine-5)-methyltransferase 1 (DNMT1; decreased in DM1 fibroblasts): DNA (cytosine-5)-methyltransferase 1, encoded by the DNMT1 gene, maintains DNA methylation profiles after DNA replication and cell division [51]. Koh and colleagues linked DNMT1 to GSK3β in fibroblasts: they found that when GSK3β is inactivated by phosphorylation, DNMT1 levels are elevated by TGF-beta1 [52]. Recent evidence shows that DNMT11 is required for correct myogenesis, a process impaired in DM1, leading to defective muscle regeneration and to muscle weakness and wasting [53, 54]. Studies performed in mice with muscle-specific deletion of Dnmt1 rev-ealed a reduced ability of myoblasts to differentiate [55]. Dnmt1-knock down in HT1080 cells, a human fibrosarcoma cell line carrying a copy of a human HPRT minigene inactivated by a (CAG)95 repeat tract, destabilizes CAG-repeats, suggesting a fundamental role of Dnmt1 in CAG-repeat instability [56].
DNMT1 expression is more pronounced in “young” human fibroblasts than in passage-aged, and DNMT11-knockdown induces a senescence pheno-type in “young” fibroblasts [57].The premature aging in DM1 patients [58] could link to the decreased DNMT1 protein level in DM1 patient derived fibroblasts (Table 1), presumably caused by GSK3β-overactivation.
Catenin beta-1 (CTNNB1; decreased in DM1 fib-roblasts): is a key component of the canonical Wnt signaling pathway controlling physiological processes like embryonic development, cellular proli-feration and differentiation [59]. In the absence of Wnt ligands, β-catenin is part of a cytoplasmic comp-lex also containing GSK3β. This complex facilitates the phosphorylation and ubiquitination of CTNNB1, thus controlling its activation and proteasome-mediated degradation. In skeletal muscle, Wnt/β-catenin signalling plays a central role in myogenesis, differentiation and myotube hypertrophy [60–62]. Consequently, CTNNB1 is considered a promising target in diseases such as cachexia or muscular dystrophies, characterized by muscle wasting. A chronic overactivation of Wnt pathways was found in skeletal muscles of DMD-patients, driving the activation of TGFβ2 and thus promoting fibrosis [63]. Of note, exogenous inhibition of the Wnt pathway was shown to reduce fibrosis in dystrophic muscles of the mdx mouse model [64]. In the disease etiology of DM1, CTNNB1-upregulation (via the Wnt signaling pathway) was linked to increased cancer risk [65]. We identified decreased CTNNB1-levels in fibroblasts of both mild and severely affected patients, and in accordance with the expression level of GSK3β, this effect is more pronounced in mild patients rather than severe.
Heterogeneous nuclear ribonucleoprotein H3(hnRNPH3; decreased in DM1 fibroblasts): is an RNA binding protein involved in splicing, in particular by participating in early heat shock-induced splicing arrest [66]. Inhibition of GSK3β controls cellular levels of hnRNPH3, thus showing a functional inter-dependence of both proteins [67]. hnRNPH3 binds to splice regulator muscleblind 1 variants (MBNL1CUG) that co-localize with CUG foci in DM1-pat-ient-derived muscle cells [68]. Furthermore, hnRNPH3 levels are increased on average 3-fold in DM1 myoblasts [68], however, this is in contradiction with the decreased level observed in our study. This might be related to cell-type specific protein-composition of these foci, perhaps in turn influenced by cell type specific expression patterns of the related proteins.
Voltage-dependent anion-selective channel protein 2 (VDAC2; decreased in DM1 fibroblasts): localizes to the mitochondrial outer membrane where it constitutes a channel for the membrane transport of various substrates [69]. GSK3β is a major positive regulator of mitochondrial permeability during oxidative stress, a trigger of apoptosis, and interacts with VDAC2 [70]. Apart from its role in regulating the flux across the mitochondrial outer membrane, VDAC2 has a fundamental function in sequestering the proapoptotic protein BAK in the mitochondrial outer membrane and maintaining it in the inactive state [71].
Mitochondrial dysfunction and impaired oxidative metabolism are known pathophysiological mec-hanisms in DM1 [72, 73]. Impaired mitochondrial bioenergetics, accompanied by complex 1-dysfun-ction and decreased VDAC2 expression, were des-cribed in quadriceps and gastrocnemius muscle of mdx mice, an animal model for Duchenne muscular dystrophy [74]. Hence, one might assume that the VDAC2-decrease in DM1 contributes to mitochondrial dysfunction and the initiation of apoptosis.
Calpain-1 catalytic subunit (CAPN1; increased in DM1 fibroblasts): also called μ-calpain, belongs to a family of Ca2 +-dependent cysteine proteases [75]. It is expressed ubiquitously as a heterodimer [76]. Calpains are involved in several physiological functions such as cytoskeletal remodeling [77], signal transduction [78], apoptosis [79], cell cycle [80], regulation of gene expression [81] and long-term potentiation [82]. Truncation and activation of GSK3β is modulated by CAPN1 [83] and recent evidence showed that GSK3β phosphorylates desmin filaments (an essential event for myofibril destruction) in a CAPN1 dependent manner [84]. In skeletal muscle, increasedcalpain activation was described in atrophic conditions following disuse, denervation, glucocorticoid treatment and sepsis [85–87] as well as in necrotic fibers from mdx mice [88]. Moreover, elevated CAPN1 levels were found in sera samples of young DMD patients not yet treated with glucocorticoids, who also present with high levels of GSK3β that decline with aging [89]. Further evidence for a significant role of CAPN1 in muscle fibre integrity is given by in vivo studies: reduced muscle necrosis and, consequently, a significantly decreased regeneration was observed in mdx mice crossed with transgenic animals overexpressing calpastatin, a CAPN1-inhibitor [90]. Based on these findings, one might suppose that increased CAPN1 levels may be linked to the dystrophic conditions characterizing DM1. Indeed, immunohistochemical studies of CAPN1 on cardiac and tibialis anterior muscles derived from DMSXL mice, a suitable DM1 mouse model, revealed an increase compared to wildtype littermates in both tissues. This molecular finding suggests an applicability of our proteomic findings on tissues of major vulnerability in DM1 such as cardiac and skeletal muscle.
CTP synthase 1 (CTPS1; increased in DM1 fibroblasts): is responsible for de novo synthesis of CTP (cytidine triphosphate) and biosynthesis of phospholipids, nucleic acids and in sustaining the proliferation of activated lymphocytes [91–93]. GSK3β is a known regulator of CTPS1 activity [94]. We hypothesize that the increased level of CTPS1 is linked to the altera-tions in lipid composition or metabolism that can occur during skeletal and cardiac myopathies [95]. With regards to the insulin resistance in the cardiac and skeletal muscles of DM1 patients and animal models (see below), it is worth noting that this not only results in increased circulating glucose but also lipid levels [96]. Thus, an interdependent co-regulation of both CTPS1 and NR3C1 in a GSK3β-dependent manner might be plausible.
Glucocorticoid receptor (NR3C1; increased in DM1 fibroblasts): is a member of the superfamily of nuclear receptors (acting as modulators of gene transcription) that, in the absence of glucocorticoid/cortisol, localizes to the cytosol in a chaperone-containing multiprotein complex [97]. GSK3β is involved in ligand-dependent activation of transcription and cellular localization of this receptor [98]. A study performed in a large cohort of DM1 patients revealed alterations in glucocorticoid metabolism with an increased reactivation of cortisone to cortisol, mainly in male patients [99]. An excess of glucocorticoids leads to insulin resistance, which is also present in 5– 17% of DM1 cases [100, 101]. In skeletal muscle, insulin resistance leads to impaired glucose uptake into the muscle fibers where it is required as an energy source. Due to insulin resistance and subsequent lipid metabolism alterations, DM1 patients can present with cardiac and vascular defects [101]. Therefore, increased NR3C1 might serve as a marker for the presence of DM1.
Histone deacetylase 2 (HDAC2; increased in DM1 fibroblasts): is a chromatin modifying enzyme that prevents gene-transcription by removal of acetyl groups from histones and deacetylates proteins such as transcription factors and those involved in the control of cell growth, differentiation and apoptosis [102]. HDAC2 expression and function is known to be controlled by GSK3β [103, 104]. In the context of DM1 pathophysiology it is important to note that two small molecule HDAC inhibitors, ISOX and vorinostat, increase MBNL1 expression, partially rescuing aberrant splicing in DM1 patient-derived fibroblasts [105]. HDAC2 might represent a marker protein in DM1-patient derived fibroblasts that plays a direct role in DM1 pathophysiology.
Dual specificity mitogen-activated protein kinase kinase 2 (MAP2K2; increased in DM1 fibroblasts): also known as MEK2, is a ubiquitously expressed protein that catalyses the phosphorylation of tyrosine and threonine in target proteins participating in the RAS– RAF– MEK– ERK signal transduction cascade. Consequently, via this cascade, MEK2 is involved in the regulation of a variety of processes including apoptosis, cell cycle progression, cell migration, differentiation, metabolism, and proliferation [106]. It also modulates phosphorylation and thus function of GSK3β via ERK [34]. Of particular interest for the muscle vulnerability of DM1, the MEK– ERK pathway is important in muscle cell differentiation [107]. Sustained activation of this pathway presumably also includes MEK2, as found in DM1 myoblasts, and can be considered as a contributor to the abnormal myoblast differentiation observed [108]. Moreover, RAS-ERK pathway activation is known to regulate alternative splicing [109], the main pathophysiological process in DM1. One of the most common symptoms in DM1, myotonia, is based on aberrant splicing of the skeletal muscle chloride channel (CLCN1) transcript [110, 111] and RAS pathway inhibitors correct this alternative splicing, alleviating some DM1 features in mice [112].
Although a transfer of the relevance of the proteomic data acquired from human skin fibroblasts could be demonstrated for CAPN1 in skeletal muscle utilizing a DM1 mouse model, further biochemical investigations would be needed to draw the same conclusion for the other marker proteins highlighted in this study. Along this line, confirmational studies on fibroblasts derived from further DM1 patients would be needed to substantiate the robustness of the proposed cellular marker proteins.
CONCLUSIONS
Overall, in this study we have identified nine proteins associated with GSK3β that may provide much-needed biomarkers for DM1 disease identification using easily obtained patient fibroblasts. Furthermore, we have also identified three proteins (CTPS1, CAPN1and HDAC2) that increase with severity of the disease, providing potential biomarkers that can be used in categorizing patients, monitoring disease progression and response to treatment. Validation of such biomarkers and their hypothesized roles in DM1 and GSK3β-associated pathophysiology will be required in future studies. Given that recent clinical trials in DM1 are based on GSK3β-inhibition [113], as the proteins defined in our study, due to their interaction with GSK3β, may serve as suitable markers to monitor success of the tested treatment concepts.
FUNDING
Financial support by the Ministerium für Innovation, Wissenschaft und Forschung des Landes Nordrhein-Westfalen, the Senatsverwaltung für Wirt-schaft, Technologie und Forschung des Landes Berlin and the Bundesministerium für Bildung und Forschung is gratefully acknowledged. This work was also supported by an unconditional grant from AMO pharma as well as a grant from the French Muscular Dystrophy Association (AFM-Téléthon; #21466) to AR. HL receives support from the Canadian Institutes of Health Research (Foundation Grant FDN-167281), the Canadian Institutes of Health Research and Muscular Dystrophy Canada (Network Catalyst Grant for NMD4C), the Canada Foundation for Innovation (CFI-JELF 38412), and the Canada Research Chairs program (Canada Research Chair in Neuromuscular Genomics and Health, 950-232279).
Footnotes
ACKNOWLEDGMENTS
We thank Mr. Daniel Cox (MRC biobank Newcastle) for expert technical assistance.
CONFLICTS OF INTEREST
All authors declare no conflict of interest.
AUTHORS’ CONTRIBUTIONS
AR, DH and HL designed the study. NN collected skin biopsies and provided clinical information. DH performed proteomic studies. AH performed data analysis and in silico studies. VG, EOC, USS, HL & AR drafted the manuscript. GG provided the tissue of the DM1 mouse model. All authors declare that the work described has not been published before and is also not under consideration for publication anywhere else. The final manuscript-draft has been approved by all co-authors.