
Abstract
Although DNA-sequencing is the most effective procedure to achieve a molecular diagnosis in genetic diseases, complementary RNA analyses are often required.
Reverse-Transcription polymerase chain reaction (RT-PCR) is still a valuable option when the clinical phenotype and/or available DNA-test results address the diagnosis toward a gene of interest or when the splicing effect of a single variant needs to be assessed.
We use Single-Molecule Real-Time sequencing to detect and characterize splicing defects and single nucleotide variants in well-known disease genes (DMD, NF1, TTN). After proper optimization, the procedure could be used in the diagnostic setting, simplifying the workflow of cDNA analysis.
DNA sequencing is the most commonly used and effective strategy for the routine diagnostics of genetically inherited diseases [1–3]. With the introduction of the next-generation sequencing platforms over ten years ago, clinical sequencing has become a routine approach [1, 2]. Targeted, exome and genome sequencing have contributed to increase the diagnostic rate of a large number of rare diseases [1–4]. Variants predicted as being splice-disrupting need to be characterized using RNA from the tissue of interest [5, 6]. Usually, a reverse transcription-polymerase chain reaction (RT-PCR) is performed using primers amplifying the region of interest [7]. In case of a splice defect caused by a heterozygous variant, RT-PCR products are often sequenced after gel extraction or after cloning them in plasmids. The procedure is even more complex when the variant occurs in regions undergoing alternative splicing or when a single variant causes multiple mis-splicing events.
On the other hand, over 50% of patients remain undiagnosed after DNA sequencing [3]. Some of them carry variants that are not identified or not properly interpreted at the DNA-level [3, 9]. RNA sequencing or cDNA analysis is an effective second-tier test, when the tissue of interest is easily accessible [8, 11]. Usually, RNA sequencing is performed to globally evaluate the transcripts and search for an outlier in a gene-independent way [8, 10]. Alternatively, the sole transcript of interest can be analyzed when its coding gene is strongly associated with the observed phenotype and DNA analysis (usually focused on the coding region) has not identified any causative variant. For example, in patients with a clinical suspicion of Duchenne muscular dystrophy, when dystrophin expression is abnormal/absent and no mutation in the DMD gene is found with MLPA or DNA sequencing, the transcript needs to be analyzed [12, 13]. Multiple RT-PCRs, followed by a direct Sanger sequencing on purified RT-PCR products, are performed to identify possible splicing defects [14].
Our study aims to verify if RT-PCR products can be pooled for library preparation and analyzed with a long-read sequencer (PacBio-RS-II) to identify and characterize single nucleotide variants and splicing defects.
We extracted RNA from the muscle biopsies of four patients with a Duchenne/Becker muscular dystrophy (DMD/BMD) using the TRIzol reagent (Invitrogen, CA, USA). Similarly, RNA from the muscle biopsies of four patients with bi-allelic causative variants in the titin (TTN) gene was extracted using the QIAGEN RN easy Plus Universal Mini Kit (Qiagen, Germany). Finally, we used the PAX gene Blood RNA Kit (Qiagen) to extract RNA from the blood of two patients with a neurofibromatosis type-I, caused by heterozygous variants in the NF1 gene.
For all the samples, 2 mg of total mRNA was converted into cDNA with the Super Script III kit (Invitrogen). The cDNA of the DMD/BMD patients and of the NF1 patients was analyzed by RT-PCR using overlapping primers spanning the entire coding sequence [12, 15]. For titinopathy patients, the TTN cDNA was amplified with specific primers to characterize the effect of five variants in canonical splice sites, previously identified by a DNA-sequencing method [16]. The RT-PCR products were then analyzed with a traditional Sanger sequencing approach (after gel extraction and/or cloning of the fragments) and a Single-Molecule Real-Time (SMRT)-Sequencing based strategy (Fig. 1).
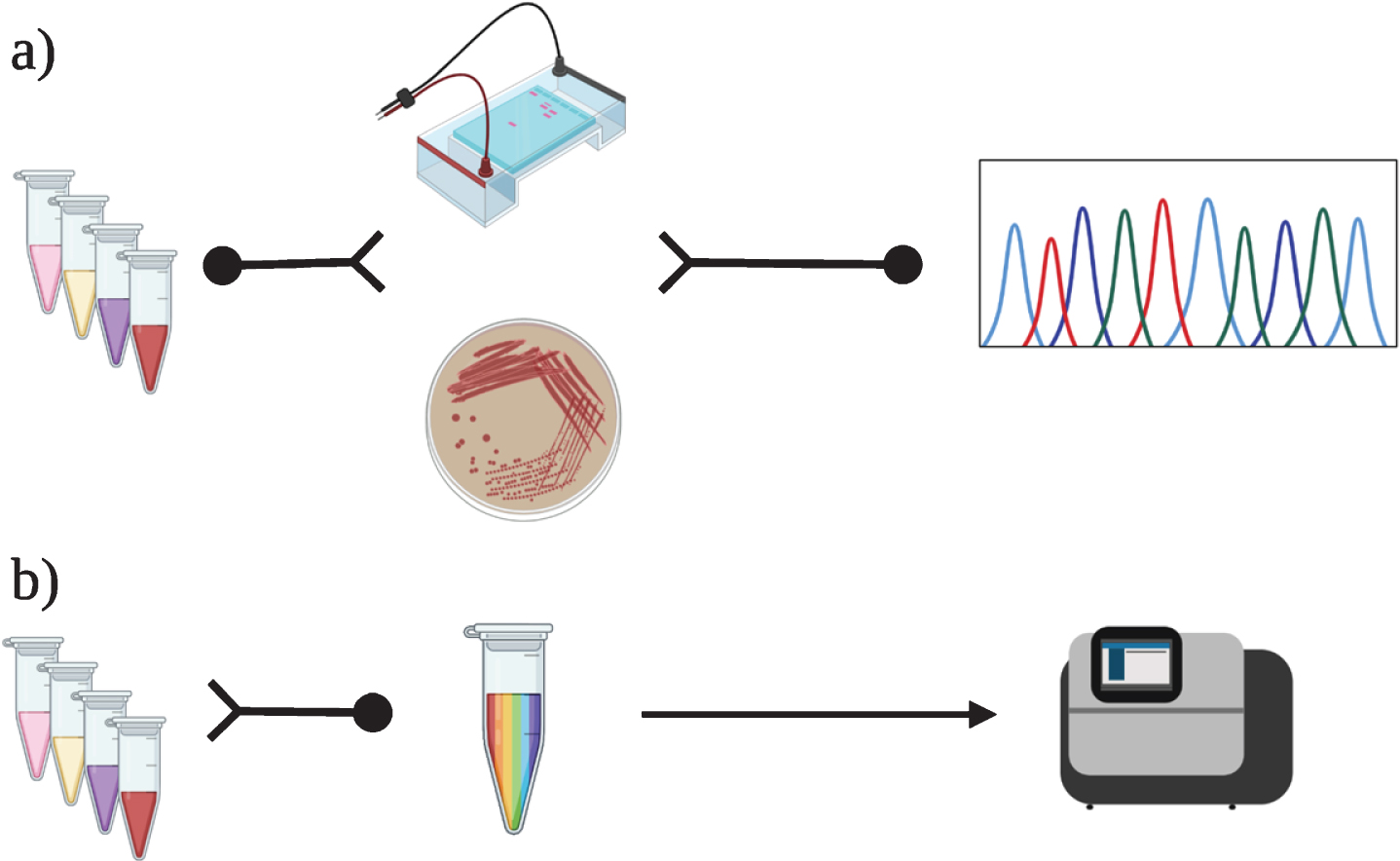
Traditional and SMRT-based cDNA analysis for splicing characterization. a) Splicing variants are traditionally identified and characterized using a reverse transcriptase-polymerase chain reaction (RT-PCR), followed by a direct Sanger sequencing on purified products. In case of multiple RT-PCR products, this requires a gel extraction or cloning of PCR products into a plasmid for sequencing. b) In the SMRT-based approach, RT-PCR products spanning the mutated regions were pooled in equimolar amounts in a single tube and the pool was used for library preparation and sequencing following the PacBio-RS-II protocol.
For the SMRT evaluation, all the RT-PCR products spanning the mutated regions were pooled in equimolar amounts. Library preparation and sequencing were performed according to the PacBio-RS-II protocol using P6/C4 chemistry with a prior size selection step to remove small library fragments (less than 700 bp). A total of 1860 Mbp of sequence was obtained from one SMRT cell. Circular consensus sequencing (CCS) reads were generated using SMRT portal 2.3 RS_Reads Of Insert protocol at least 8 full-pass sub reads.
The traditional sequencing revealed the disease-causing variants in all the patients. As summarized in Table 1, the SMRT-based approach confirmed the results obtained by the Sanger-based cDNA analysis, characterizing the splicing defects in the DMD transcripts, due to hemizygotic variants. Similarly, in patient NF1_1, the SMRT-analysis identified both the wild type transcript and the mutated one showing the skipping of exon 12. In patient NF1_2, the heterozygous single nucleotide variant c.5426 G > T resulting in a missense variant was detected by both approaches. The variant c.5426 G > T p.(Arg1809Cys) has been previously identified in several families with a mild form of neurofibromatosis and it is currently listed as pathogenic in ClinVar [17, 18]. In patient TTN_1, both methods identified the complete skipping of exon 67; in patient TTN_2 the insertion in the transcript of the first intronic nucleotide of intron 362; and in patient TTN_4 the partial skipping of exon 26. In patient TTN_3, the variant c.15776-1G>T caused the intron 54 retention. The second variant (c.67349-2A>C), interestingly, caused a partial skipping of exon 320 and the insertion in the transcript of the intron 319.
Variants identified and characterized
Genomic coordinates are in genome assembly GRCh37 (hg19). Reference transcripts: DMD (NM_004006.1), NF1 (NM_000267.3), TTN (NM_001267550.1).
Long-read sequencing is increasingly used in clinical sequencing applications since it is expected to identify possible disease-causing variants usually not detected by short-read technologies (e.g. structural variants and tandem-repeats) [19–21]. This preliminary study demonstrates that the SMRT sequencing is also useful for characterizing splicing defects.
A comprehensive molecular analysis, including DNA and RNA tests, is often essential for a correct diagnosis and a proper evaluation of prognosis. As seen in patient DMD_1, single nucleotide variants can be misinterpreted as missense changes although a secondary RNA analysis can reveal a deleterious effect on the splicing.
Similarly, downstream effects of splicing variants identified at the DNA-level should be carefully evaluated to finely map the consequences on the transcript(s) and, thereby, on the protein [5]. However, sometimes, the characterization of splicing defects can be tedious and expensive. Our data suggests that pooling RT-PCR products in equimolar amounts and analyzing them with a long-read sequencer, without performing any complex and time-consuming post-PCR methodology (gel extraction or cloning), is a successful method to analyze splice aberrations.
On the other hand, when there is a specific clinical suspicion and DNA tests have not identified any causative genetic variant, analysis of the cDNA of interest is still a useful second-tier test. One percent of patients with a DMD phenotype, for example, carry splicing variants only identified with a cDNA analysis [12]. Similarly, cDNA analysis is a valuable second-level test for patients with a clinical diagnosis of NF1(15), or with a suspected recessive titinopathy, when only a monoallelic truncating variant has been identified [6, 22]. Analyzing the full-length cDNA, especially for large genes, is complex and not easily scalable [23]. Traditionally, for RNA analysis of the entire coding sequences, primer pairs that amplify partially overlapping fragments of 500–700 bp are designed [15]. RT-PCR products are thereby first analyzed by agarose gel electrophoresis and subsequently by bidirectional Sanger sequencing [15].
A simplified approach could be implemented using a SMRT-based strategy. The transcript of interest could be amplified through multiple long RT-PCRs (5–10 Kb), thereby, using a reduced number of primers and performing a reduced number of PCRs. Optimizing the technical aspects (primer/PCR design and equimolar pooling), RT-PCR products could be pooled and run with a PacBio sequencer, further simplifying the transcript analysis.
Footnotes
ACKNOWLEDGMENTS
The authors would like to thank for the support: Magnus Ehrnrooth Foundation (MS), PäivikkijaSakariSohlberginSäätiö (MS and MJ), PaulönSäätiö (MS), Biomedicum Helsinki säätiö (MJ), Jane and Aatos Erkko Foundation (PH and BU), MedicinskaUnderstödsföreningen Liv ochHälsarf (PH), Folkhälsan Research Foundation (BU), Juselius Foundation (BU), Academy of Finland (BU). We want to thank Kirsi Lipponen for Pacbio sequencing.
Figure 1 was created with BioRender (![]() ).
).