
Abstract
Background:
Biallelic variants in Anoctamin 5 (ANO5) gene are causative of limb-girdle muscular dystrophy (LGMD) R12 anoctamin5-related, non-dysferlin Miyoshi-like distal myopathy (MMD3), and asymptomatic hyperCKemia.
Objective:
To describe clinic, histologic, genetic and imaging features, of ANO5 mutated patients.
Methods:
Five patients, four from France (P1, P2, P3 and P4) and one from Mexico (P5), from four families were included. P1 and P2, belonging to group 1, had normal muscle strength; Group 2, P3, P4 and P5, presented with muscular weakness. Muscle strength was measured by manual muscle testing, Medical Research Council (MRC) grades 1/5 to 5/5. Laboratory exams included serum CK levels, nerve conduction studies (NCS)/needle electromyography (EMG), pulmonary function tests, EKG and cardiac ultrasound. ANO5 molecular screening was performed with different approaches.
Results:
Group 1 patients showed myalgias with hyperCKemia or isolated hyperCKemia. Group 2 patients presented with limb-girdle or proximo-distal muscular weakness. Serum CK levels ranged from 897 to 5000 UI/L. Muscle biopsy analysis in P4 and P5 showed subsarcolemmal mitochondrial aggregates. Electron microscopy confirmed mitochondrial proliferation and revealed discontinuity of the sarcolemmal membrane. Muscle MRI showed asymmetrical fibro-fatty substitution predominant in the lower limbs.
P1 and P2 were compound heterozygous for c.191dupA (p.Asn64Lysfs*15) and c.1898 + G>A; P3 was homozygous for the c.692G>T. (p.Gly231Val); P4 harbored a novel biallelic homozygous exons 1–7 ANO5 gene deletion, and P5 was homozygous for a c.172 C > T (p.(Arg 58 Trp)) ANO5 pathogenic variant.
Conclusions:
Our cohort confirms the wide clinical variability and enlarge the genetic spectrum of ANO5-related myopathies.
INTRODUCTION
Anoctamins, also known as transmembrane protein 16E (ANO/TMEM16), are a family of transmembrane proteins that comprises at least ten members, many of which have been shown to correspond to calcium-activated chloride channels [1, 2]. These proteins are involved in a variety of functions such as phospholipid scrambling, membrane excitability in cardiac muscle and neurons, regulation of vascular tone, and photoreception [1–5]. The anoctamin-5 gene (ANO5; #OMIM608662), contains 22 exons and encodes a 913-aminoacid protein [4, 7]. ANO5 is highly expressed in cardiac and skeletal muscle, chondrocytes, and osteoblasts, suggesting its important role in the musculoskeletal system [5, 8–10].
Pathogenic ANO5 variants have been associated with autosomal dominant gnathodiaphyseal dysplasia (GDD; #OMIM 166260) [7], non-dysferlin Myoshi muscular dystrophy (MMD3; #OMIM 613319) [11], autosomal recessive limb-girdle muscular dystrophy anoctamin5-related (LGMDR12; #OMIM611307) [11, 12], and isolated hyperCKemia [13]. To date, more than 200 ANO5 pathogenic variants have been reported, without a clear phenotype/genotype correlation (https://databases.lovd.nl/shared/variants/ANO5) [14, 15].
The reported prevalence of ANO5 pathogenic variant ranges from 2–25% [13, 15–21]. The pathogenic variant c.191dupA in exon 5 (p.Asn64Lysfs*15) is common in Northern Europeans, and it is probably due to a founder effect [22]. In a cohort of 38 French ANO5 patients, sixteen harbored the c.191dupA [23].
Muscle histopathology findings in ANO5-related myopathies are variable, ranging from mild dystrophic features to unspecific findings [23–26]. Immunohistochemical and Western Blot studies are not useful due to the lack of an efficient commercial antibody able to detect ANO5 absence/reduction [27].
In this paper, we describe five patients with variable muscle involvement caused by pathogenic variants in ANO5. We report hereby a novel large biallelic ANO5 deletion and confirm the ultrastructural sarcolemmal membrane discontinuity in two patients.
PATIENTS AND METHODS
Four patients from France (P1, P2, P3 and P4) and one from Mexico (P5) were examined at Paris-Nord/Est reference Center of Neuromuscular Diseases, Raymond Poincaré Hospital, Paris, France, and at the National Institute of Rehabilitation Luis Guillermo Ibarra Ibarra, Mexico City, Mexico. The age of onset was variable, ranging from ten to eighty-five years old. Clinical data were systematically retrieved and retrospectively analyzed. Patients were grouped in two groups: Group 1 included patients with normal muscular strength (P1 and P2); Group 2 comprised patients with muscular weakness (P3, P4 and P5). Muscle strength was measured using the 5-scale grading manual muscle testing of the Medical Research Council (MRC). Laboratory exams included serum CK levels, nerve conduction studies (NCS)/needle electromyography (EMG), pulmonary function tests, electrocardiogram (EKG) and cardiac ultrasound.
P3 and P5 underwent an open biopsy of the deltoid and vastus lateralis muscles, respectively. Muscle samples were processed for morphologic histochemical and ultrastructural analyses according to standard procedures [28, 29]. Whole-body muscle magnetic resonance imaging (MRI) by multi-axial technique in the axial plane (axial T1 weighted, IDEAL sequences, T2 Fat equivalent, T1 water equivalent STIR, and Dixon) was performed in all patients, except P5 who underwent lower limbs MRI.
Genetics
LGMD Nimblegen, Roche® myopathe_v3’s panel designed to screen 38 genes associated to LGMDs was performed in P1 and P2. LGMDs Ampliseq designer Life Technologies® panel designed to screen ANO5, CAPN3, CAV3, DYSF, EMD, FKRP, LMNA, SGCA, SGCB, SGCD, SGSG, and TRIM32 genes was performed in P3 and P4. In P5, a 10 gene panel including ANO5, CAP3, DYSF, FKRP, SGCA, SGCB, SGCG, TCAP and GAA genes was performed. All the variants were assessed according to the guidelines of the American College of Medical Genetics and Genomics [30].
RESULTS
Group 1
Individual clinical, laboratory and genetic data are summarized Tables 1 and 2.
Summary of patient’s clinical history and clinical presentation
ANO5 pathogenic variants identified in the patients
Prediction program: Mutation Taster, Polyphen2, or SIFT. NA: not applicable.
P1 is a 23-year-old woman, referred in her teens for exercise induced fatigability, myalgias and hyperCKemia (<1000 UI/L). Myalgias were particularly intense and invalidating. She was treated with increasing doses of opioids with scarce effect. Neurologic examination was unremarkable except for slightly hypertrophic calves.
P2, P1 younger sibling, presented asymptomatic hyperCKemia ranging between 1500 and 5000 UI/L, discovered after her sister attended to our Center.
EMG, cardiac, and respiratory workup were normal in both P1 and 2.
P1 whole body MRI failed to show any alteration. Notably there was the complete absence fat infiltration, muscle atrophy and muscle edema with T2, STIR, and IDEAL sequences. Genetic analyses showed that both siblings are compound heterozygous for the pathogenic variants c.191dupA (p.Asn64Lysfs*15) and c.1898 + G>A in ANO5 (Table 2). Unaffected parents were carrier of one pathogenic variant, each.
Group 2
Individual clinical, laboratory and genetic data are summarized Tables 1 and 2.
P3 is a 56-year-old woman. She noticed myalgias in thighs, arms, shoulder and back from age 30 years. The myalgias impaired importantly her quality of life and they were resistant to opioids. At age 45 years, she presented gait disturbances and difficulties in climbing stairs. Her neurologic examination disclosed axial, and limb girdle muscle weakness (triceps 4/4, deltoids 4/4, wrist flexion 4/4, hamstrings 4/4, quadriceps 4+/4+, gluteus medius 4/4, ilio-psoas 4/4 and gluteus maximus 4/4). Serum CK levels ranged from 1500 to 1700 UI/L.
P4 is an 88 years old man who developed progressive walking difficulties and myalgias after effort at 85 years. Physical examination disclosed asymmetric atrophy of calves, and slight scapular winging. He also had a waddling gait and cannot go on heels and tiptoes. Manual muscular testing revealed lower limbs proximo-distal weakness (hip abduction: 3/2; hip adduction: 3/3; quadriceps 4/5 and hamstrings 4/5 MCR). Serum CK levels were 897 UI/L.
P5, a 48 years old man presented lower back and lower limbs pain in his forties followed by lower limb muscular weakness provoking progressive gait difficulties. He successively lost the ability to dance and run, and developed difficulties in rising from a chair and go downstairs. His neurologic examination at age 45 revealed severe quadriceps atrophy, and proximal lower limbs weakness (iliopsoas 3/3, gluteus maximum and medium 2/2, quadriceps 2/2, hamstrings 3/3, anterior tibial 4/4 posterior tibial 4/4 MRC). Serum CK levels ranged between 1160 and 1600 UI/L.
EMG findings in group 2 were heterogeneous. An inconspicuous and asymptomatic sensitive axonal neuropathy was reported in P3, a myopathic process, with slight pseudo-neurogenic recordings was found in P4, and P5 had a myopathic process in proximal muscles.
Cardiac and respiratory workup were normal in P3 and P4; P5 showed EKG changes suggestive of chronic cardiac ischemia.
Muscle MRI finding in P3 (Fig. 1A) showed a predominant fatty replacement paraspinal and thigh muscle without remarkable bright signal on T2 fat saturated images.
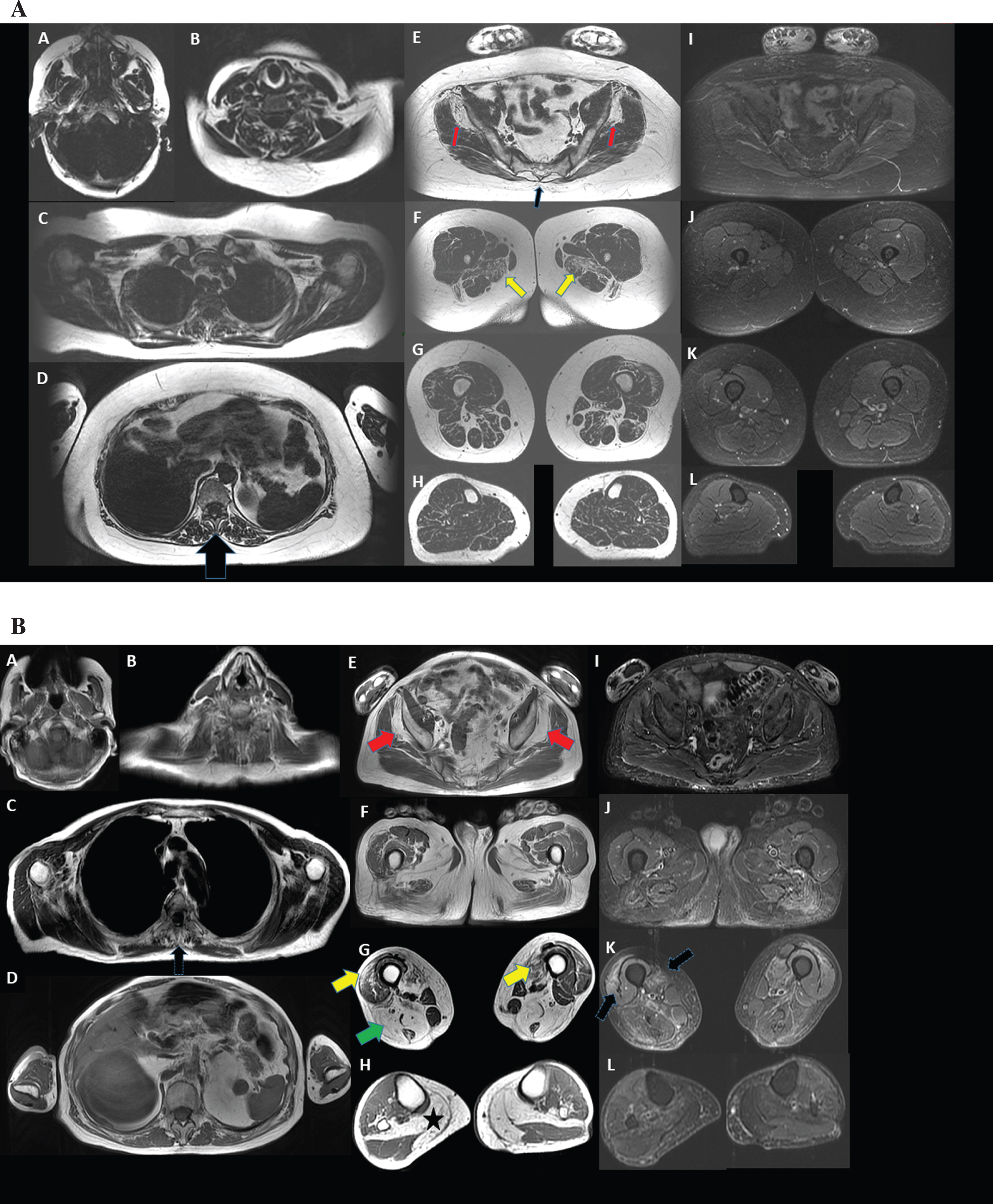
Magnetic Resonance Imaging (MRI) Selected axial TIW images of muscles. A. Patient 3. Axial T1 weighted sections from head to toes (fat images of the Dixon T2 examination, A to H) and axial sections T2 fat saturated from pelvic girdle to toes (Water images of the Dixon T2 examination, I to L). The muscles of the face and upper limbs are well preserved and do not show any bright signal of fatty replacement on T1 or “inflammation” on T2 fat sat. There is a predominant fatty replacement on bright signal on T1 in erector spinae (D, indicated by the black arrow), gluteus minimus (E, indicated by the red arrows), and adductor magnus (F, indicated by the yellow arrows). There is no remarkable bright signal on T2 fat saturated images. B. Patient 4. Axial T1 weighted sections from head to toes (A to H) and axial sections T2 STIR from pelvic girdle to toes (I to L). Muscles of the face and upper limbs are well preserved and do not show any bright signal of fatty replacement on T1 or “inflammation” on T2 STIR. There is a predominant fatty replacement of bright signal on T1 in erector spinae (C, indicated by a black arrow), gluteus minimus, adductor longus and magnus, vastus lateralis and medialis, more visible in distal segments (E, indicated by the red arrow and G, indicated by the yellow arrows), hamstring (G, indicated by a green arrow), medial gastrocnemius and soleus (H, indicated by the black star). The involvement is symmetric. There is remarkable bright signal on T2 STIR images in vastus medialis and lateralis (K, indicated by the black arrows).
P4 muscle MRI (Fig. 1B) showed a predominant and symmetric fatty replacement in spinal, thigh and leg muscles (Fig. 1B) and a remarkable bright signal on T2 STIR images was present in vastus medialis and lateralis (K, indicated by the black arrows).
Lower limbs MRI of P5 showed an asymmetric fibro-fatty replacement of posterior compartment of thighs, with a more selective atrophy of the semimembranosus, semitendinosus, biceps femoris, flexor hallucis longus, flexor digitorum longus, tibialis posterior, medial gastrocnemius, soleus and popliteus muscles (not shown).
Genetic studies revealed a homozygous c.692G>T. (p.Gly231Val) pathogenic variant in P3.
P4 harbored a novel biallelic homozygous deletion of exons 1 to 7. P5 was homozygous for the already reported c.172 C > T (p.Arg 58 Trp) pathogenic variant.
A deltoid and vastus lateralis muscle biopsies were performed in P3 and P5, at 50 and 41 years, respectively. Both biopsies failed to show dystrophic features. In P3, hematoxylin and eosin staining (H&E) revealed rare nuclear internalizations (Fig. 2A), and modified Gömöri staining (mGT) disclosed the presence of a rim of fuchsinophilic material corresponding to increased subsarcolemmal mitochondria in approximately 70% of fibers (Fig. 2B). The muscle biopsy from P5 disclosed the presence of prominent subsarcolemmal mitochondrial aggregates visible with both mTG and hystoenzymatic oxidative reactions (Fig. 2C). Electron microscopy studies in both patients confirmed the presence of subsarcolemmal mitochondrial proliferation (Fig. 2D, and E), as well as subsarcolemmal membrane discontinuities (Fig. 2F).

A and B. Light microscopy of deltoid muscle biopsy from P3 performed at 50 years. A. H&E staining showing normal morphologic features; B. mTG staining showing the presence of fuchsinophilic subsarcolemmal crescents corresponding to accumulated mitochondria; C. NADH staining from P5 showing the presence subsarcolemmal mitochondrial aggregates. D, E and F Electron micrographs from P3 and P5; D. Electron micrograph showing the presence of numerous subsarcolemmal mitochondria of normal shape and size (indicated by an asterisk); E. Presence of a large intracytoplasmic and subsarcolemmal mitochondrial proliferation; F. Presence of areas of membrane discontinuity (indicated by the arrows) suggesting a damage of the plasma membrane.
DISCUSSION
We presented the clinical and laboratory findings of five patients with ANO5 pathogenic variants and variable muscular phenotypes.
In our cohort, we observed that only one of the three female patients (P3) presented evident muscular weakness, while both male, P4 and P5, showed a full-blown LGMDR12 phenotype.
Group 1 patients are compound heterozygous for two pathogenic variants in ANO5 (c.191dupA and c.1898 + 1G>A). Remarkably, P1 experienced muscular fatigability and P2 was studied due to the discovery of elevated serum CK in her sister. This finding suggests that ANO5-related myopathies shows a wide clinical spectrum and that might be underdiagnosed in patients with oligosymptomatic phenotypes or with isolated hyperCKemia [12, 15]. In group 2, the full LGMDR12 phenotype was observed in 2 cases (P4 and P5), while the MMD3 phenotype was observed in P3. Our findings confirm that males have a tendency to develop a more severe phenotype in contrast with females, who are frequently oligosymptomatic or asymptomatic [15, 30]
In our cohort, the age of onset was highly variable, and EMG studies showed variable electrophysiological changes. Regarding the cardiac and respiratory involvement, we confirmed the absence of alterations [11, 30]. Muscular pain was experienced in three cases (P1, P3 and P5). Myalgias, isolated or associated to muscular weakness, has been already described in ANO5-related myopathies [26, 30]. However, the pathophysiology of muscular pain related to ANO5 has not been elucidated. Noticeably, P1 and P3 experienced a consistent pain that impaired their quality of life, and opioid treatment only lead to scarce relief. Myalgias has been associated to other LGMDs such as Calpain-related muscular dystrophies, LGMDR1, (personal observation), facioscapulohumeral muscular dystrophy 1 (FSHD1), and other rare conditions [31–33].
Whole body MRI is very useful to assess the muscular damage, and to determine the progression of the muscle disease in LGMDs [33]. Asymmetric muscle involvement is frequently observed in ANO5 patients [34]. MRI in multi-axial technique in the axial plane in IDEAL sequences T2 provide the information to evaluate myopathies in order to detect fat infiltration, muscle atrophy and muscle edema [35]. In LGMDR12, the most common pattern is mild to moderate involvement of the gluteal muscles, major involvement of the muscles of the posterior compartments of the thighs and calves, and a patchy involvement of the anterior thigh compartment; the adductors, semimembranosus, semitendinosus, gastrocnemius and soleus are the most severely affected muscles [34, 36]. P3 and P4 whole body muscle MRI findings confirmed the above-mentioned findings, although mainly symmetric. In P4 we also noticed an involvement of erector spinae and a remarkably bright signal in TE STIR images in vastus lateralis and medialis muscles. Even if we weren’t able to demonstrate MRI changes in the group with normal muscular strength, it is important to perform a full MRI study including T1-weighted, short-tau inversion recovery (STIR) or the multi-axial technique in the axial plane in IDEAL sequences T2, in order to observed incipient muscular changes [35].
The yield of muscle biopsy in the diagnostic algorithm of LGMDR is important to demonstrate the presence of a necrotic regenerating pattern, and the identification of absence/reduction of protein expression [37]. Immunohistochemical and protein studies are of relative interest in LGMDR12 due to the unavailability of trustable and sensitive commercial antibodies. Of note a Western blot approach of protein quantification has been developed [23], but this is not currently available in the diagnostic routine.
Muscle biopsies performed in P3 and P5 only showed mild myopathic alterations and subsarcolemmal mitochondrial proliferation. The latter have been also found in LGMDR2, dysferlin-related myopathies muscle samples from patients and a mouse model [38, 39].
With ultrastructural studies, we confirmed the presence of sarcolemmal membrane discontinuity (Fig. 2) possibly explaining the constant and prominent serum CK increasing in ANO5-related myopathies [11].
From genetic standpoint, P1 and P2, harbored the common c.191dupA, and the already reported c.1898 + 1G>A intronic variant affecting a splicing donor residue [16, 40]. The c.191dupA is the most common pathogenic variant in Northern European population and it is probably related to a founder effect.
P3 was homozygous for the previously reported c.692G>T pathogenic variant [11, 23].
P4 harbored an unreported biallelic deletion involving exons 1–7. Only two cases with large deletions in ANO5 have been reported, one of them involving exons 13–17, and the other being a big 418 kb deletion including the entire ANO5 gene [24, 26]. It was not possible to perform a gene-centric array comparative genomic hybridization (aCGH) to determine the exact genomic coordinates. Therefore, it was not possible to perform the subsequent sequence analysis of the breakpoint regions. In both previously reported ANO5-related myopathies associated to large deletions, the result was an out - of - frame ANO5 transcript with a consequent not functional protein product [24, 26]. Due to the exons involved in the deletion of our patient, and the characteristics of the altered nucleotide sequence, we can assume that the change occurs in the absence of a functional protein. Recent studies have shown the relevance to search for copy number variation (CNV) in those cases in which was not possible to detect a single nucleotide change that could explain a neuromuscular phenotype [41]. Of note. Thomas et al, showed that 3,051 out of 32,590 patients suspected of a neuromuscular disorder harbored CNVs. Of these 3,051 cases, 1, 328 were associated to autosomal recessive conditions, and 856 at homozygous state [42]. This finding suggests the importance to determine the copy number variations in order to identify mutation responsible of ANO5 related conditions [24, 26].
In conclusions, our study confirms the high variability of muscle phenotype and enlarge the genetic spectrum of ANO5-related muscle myopathies.
FUNDING
Conflict of interests
The authors declare that they have no conflicts of interest.
Footnotes
ACKNOWLEDGMENTS
We are thankful to all member of Risler lab of the Institute of Myology in Paris for technical assistance of P3 muscle biopsy.