
Abstract
Hereditary neuropathy with liability to pressure palsies (HNPP) is an autosomal dominant inherited disorder commonly presenting with acute-onset, non-painful focal sensory and motor mono neuropathy. In 80% of cases, the genetic defect is a 1.5 Mb deletion on chromosome 17p11.2, including PMP22. Only few cases of partial deletion and point mutations in PMP22 are involved in HNPP. We investigated a 62-years-old man with lower limb plexopathy first considered as Garland’s syndrome. A month later, his 29 years old son also consulted for paresthesia on the peroneal nerve.
Targeted sequencing of the PMP22 gene identified a c.370delT (p.Trp124Glyfs*31) in both affected patients.
We report a new PMP22 point mutation associated with an atypical clinical phenotype of HNPP, a painful plexopathy of the lower limb worsenen by diabetes and a mere paresthesia, but a typical ENMG. This study illustrates the large spectrum of the disease, and emphasizes the importance of a complete ENMG and family history.
HIGHLIGHTS
PMP22 direct sequencing should always be performed in the absence of the deletion Diabetic neuropathy can hide hereditary neuropathy in familial context Electrophysiology represents the keystone of the clinical diagnosis
INTRODUCTION
PMP22 related neuropathies mainly comprise PMP22 duplications leading to Charcot-Marie-Tooth disease type 1A (CMT1A), PMP22 deletions, producing Hereditary Neuropathy with liability to Pressure Palsies (HNPP), and PMP22point mutations, producing both phenotypes [1]. The pathophysiolocal mechanism for both diseases was initially described as a loss of function by haplo insufficiency for HNPP and toxic gain of function for CMT1A [2].
HNPP is characterized by recurring sensory and motor neuropathy in a single nerve starting in adolescence or young adulthood, usually at entrapment sites or susceptible pressure points, mostly targeting the ulnar or peroneal nerve. It is well established that the deficit recovers painlessly after a minor trauma [3]. Histology from nerve biopsy often displays sausage-like swellings, or “tomacula”, from the myelin sheaths.
HNPP is frequently under diagnosed or misdiagnosed owing to the heterogeneity of clinical and electrophysiological appearance, for example progressive mono neuropathies, chronic sensory neuropathy, transient sensory symptoms or Charcot-Marie-Tooth-like presentation can also be found. In the majority of cases (80%), the genetic defect consists of a recurrent deletion of the 17p11.2p12 region encompassing 1.5 Mb including the PMP22 gene [4, 5]. Some cases reported HNPP patient harboring a heterozygous partial deletion of PMP22 5’ region containing exon 1 to 3 [6]. 20% of HNPP patients carried point mutations [3]. Point mutations resulting in a frame shift or a nonsense mutation reported are summarized in Table 1, including age at onset and clinical expression [7–19].
Reported point mutations in the literature
In this study, we describe a clinical, electrophysiological and molecular genetic study of a family with a HNPP phenotype presenting a novel frame shift mutation in the exon5 of the PMP22 gene.
CASE REPORT
Clinical aspects
The proband is a 62 years old man, whose only medical record is diabetes (well balanced). The onset consisted of paresthesiain the left lower limb, numbness around the anus, loss of bladder control and a gradually motor deficit of the foot lifters, plantar flexion of the foot and at lesser extent knee extension. An MRI showed no spinal cord compression and biological analyses from peripheral blood and CSF only revealed high homocysteinemia. Moreover he presented important pain at the anterior surface of the thigh. Ankles jerk reflexes were abolished as well as left patellar reflex, the right one only being diminished. Clinical examination also revealed bilateral hypoesthesia up to mid calves apparently ancient. No upper limb deficit, dermatologic lesion or biological anomalies were present. So we found a lumbosacral plexopathy along with a sensory neuropathy.
His 29 years old son’s complain was directed towards right lower limb paresthesia without motor nor reflexes defect. Biological analyses did not reveal the presence of type 2 diabetes.
The rest of the examination was normal, the electrophysiological findings showed in both patients a decrease in the velocity of nerve conduction in different entrapment sites, both ulnar nerves, median nerve and one fibular (for the father), no sensory potential found at the lower limbs, and axonal sensory-motor loss predominant in the lower limbs the different scores are reported in Table 2.
Nerve conduction studies from the proband
Nerve conduction study of the proband: compression of the median nerves in the carpal tunnel and of the ulnar nerves at the elbow. Axonal loss with no excitable nerves at the lower limbs. Abnormal results are in bold. Antidromic technique was applied for sensory nerve conduction.
Molecular exploration
Due to the family history and the typical electrophysiological findings, research of the 1,5 Mb deletion on 17p11.2p12 responsible for HNPP was conducted. After informed consent, blood samples were obtained from both patients. Genomic DNA was extracted from peripheral blood cells using a standard protocol. Molecular analysis by Multiplex Ligation-dependent Probe Amplification (MLPA) of the PMP22 genewas performed as manufacturer recommendation (MRC-Holland©). No PMP22 deletion was found.
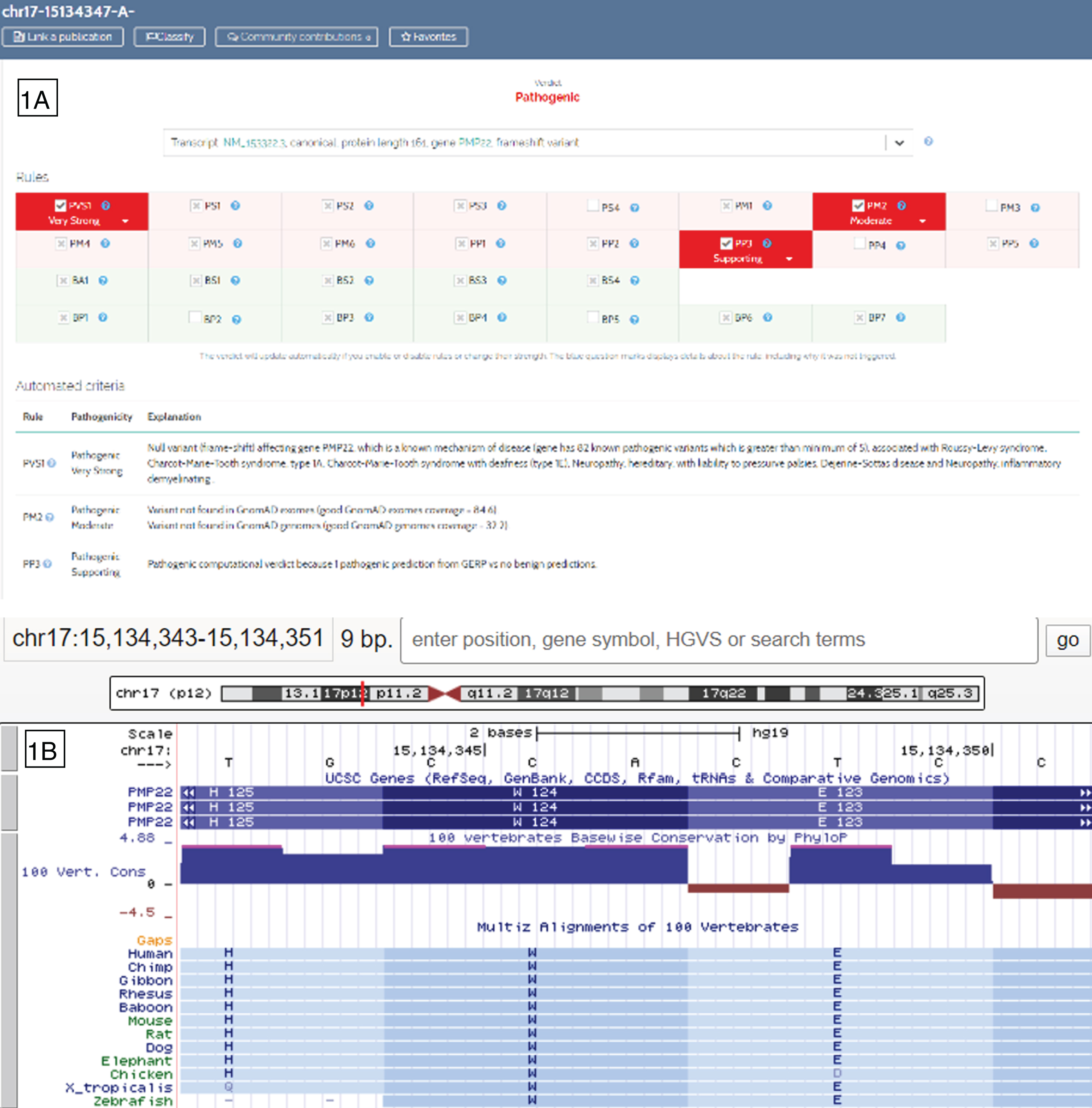
Arguments for the PMP22:c.370del pathogenicity retrieved from the Varsome website (1A) and from amino acid conservation among species (1B) shown from UCSC Genome Brower (https://genome-euro.ucsc.edu).
HNPP diagnosis was discussed. Given the high probability of clinical diagnosis and confronting with Dubourg et al. [20] guidelines for HNPP, research for PMP22 point mutations was conducted.
All coding and flanking intronic sequences of PMP22 were amplified by PCR. Sanger sequencing was then performed using the Big Dye 3.1 (Applied Biosystems©, Foster City, CA) on an ABI3500XL Life Technologies, USA(Applied Biosystems©). Amplicon sequences were compared with the reference PMP22 coding genome (hg19, NM_000304.3). A single nucleotide deletion in exon 5, c.370delT (p.Trp124Glyfs*31), was identified, leading to a truncated protein. This variant was never reported in any database (dbSNP, gnomAD). ACMG criteria met for the classification of this novel mutation were retrieved on the Varsome Website (http://www.varsome.con; Fig. 1A): Pathogenic Very Strong 1, Pathogenic Moderate 2 and Pathogenic Poor 3 were in favor of a new pathogenic variant. More precisely, PMP22 has a probability of being loss-of-function intolerant (pLI) of 0,91 supporting the haplo insufficiency by a frame shift and a premature stop cod on. Moreover, this amino acid is highly conserved among species (Fig. 1B). The genetic mechanism reflected in this truncated protein is thus identical to the more generic CNV found in HNPP patients. No other mutation was identified. The segregation analysis in the family found the same mutation for the father. Bioinformatics software were used for predicting functional impacts of the PMP22 variants as PolyPhen-2 (http://genetics.bwh.harvard.edu), Mutation Taster (http://www.mutationtaster.org), and UMD predictor (http://umd-predictor.eu/). All predictions were disease causing.
Description of variants and phenotype
Point mutation and phenotype have been submitted to LOVD database (https://databases.lovd.nl/) and can be found at the accession number #0000497671.
DISCUSSION
We described a family with a new point mutation in the PMP22 gene inherited from the affected father. This mutation has not yet been described; nonetheless Pareyson and al. had noticeda family affected by HNPP with a nonsense mutation in the same cod on in Human genomic Database Mutation (HGMD) (http://www.hgmd.cf.ac.uk/ac/search.php) but the data was unpublished [21].
Reported variants (Table 1) were clearly frame shifts and nonsense mutations leading to a truncated protein compatible with loss of function alterations leading to haplo insufficiency [2]. Few cases of reported amino acid changes seem to be associated with a milder phenotype or even clinically asymptomatic patients (c.88G>A (p.Val30Met)[10]). The new point mutation leading to a frame shift described in this report falls under the molecular spectrum of HNPP within the absence of the recurrent 17p11.2 deletion.
HNPP typically leads to episodic, painless, recurrent, focal motor and sensory peripheral neuropathy [3]. Many other phenotypes were described such as progressive mononeuropathies, chronic sensory neuropathy, transient sensory symptoms, Charcot-Marie-Tooth-like presentation, even plexopathy but only those of the upper limb were described in HNPP. Contrarily to other patients with typical HNPP, the pro band presented with important pain at the anterior surface of the left thigh, a deficit alongside with paresthesia of this member and diabetes, which initially led to the diagnostic of Garland’s syndrome. Indeed, the pain is not typical even if Yilmaz and al. [22] show that in their series of 34 patients, 75% described pain, associated with a clinical diagnosis of fibromyalgia. Moreover, even if brachial plexopathyis often described, lumbosacral plexophathy is not.
Diabetes was a confounding factor, responsible for the ankle jerk reflexes loss. No other complication was found and the disease was well-balanced. Moreover, the scan didn’t show any tumoral mass and no trauma was described. Li J and al. suggest that diabetes may have an impact on the severity of the HNPP [23]. An MRI was also made to set aside the diagnosis of compression for which sacral manifestations were absents.
The son’s pro band similar clinical presentation ruled out the diabetes complications for the neuropathy towards a dominant genetic mechanism.
The ENMG showed typical HNPP characteristics [24, 25]: polyneuropathy, median terminal motor prolongation, and focal neuropathies with compression at multiple sites, at least five. These findings first led to the research of the 17p11.2p12 1,5Mb recurrent deletion which revealed no alteration. The PMP22 gene sequence analysis was secondly performed.
The number of PMP22 point mutations responsible for HNPP is expending but many case reports are presented with a clinical heterogeneity from the 17p11.2p12 deletion phenotype, especially experiencing neuropathic pain [23]. Here we described also a new presentation of HNPP in one family with a novel point mutation in PMP22. This study also highlights the need of proper electrophysiology like the research for entrapments, even without a spoken complain, and the need to look for a domestic disorder. Therefore it seems important to expend the diagnostic for PMP22 mutations in the absence of the recurrent micro deletion.
CONFLICT OF INTEREST
On behalf of all authors, the corresponding author states that there is no conflict of interest.
AUTHOR CONTRIBUTION
BT, AS and NB took primary responsibility for the study; MN performed experimental study; KL, DE, and FE collected the data. All authors contributed to the critical revision and final approval of the version to be published.
Footnotes
Acknowledgments
The authors would like to thank the patients for allowing us to share their stories.