
Abstract
Background:
The phenotypic spectrum of the skeletal muscle voltage-gated sodium channel gene (SCN4A) mutations has been expanding dramatically with advancements in genetic testing. Previously only known to cause autosomal dominant myotonia or periodic paralysis, now recessive mutations have been found causing congenital myopathies and congenital myasthenic syndromes.
Case presentation:
A 27-year-old woman who was born with Arnold-Chiari malformation, hydrocephalus, high-arched palate, bilateral hip dysplasia, and severe scoliosis presented for evaluation of episodic muscle stiffness and weakness. Electrodiagnostic studies revealed myopathy and widespread myotonia. Muscle histopathology showed marked fiber size variability, type I fiber predominance with minimal scattered necrosis and regeneration which was typical of a congenital myopathy with an additional finding of a lobulated structural pattern in type I fibers. Sequential individual gene testing revealed a novel de novo heterozygous c.2386 C > G, p.Leu796Val missense mutation in the SCN4A gene.
Discussion:
To the best of our knowledge, this is the first report of a dominant, heterozygous mutation in SCN4A causing a complex phenotype of congenital myopathy and myotonia with multiple congenital anomalies and unique muscle pathology findings. This case is another addition to the ever expanding phenotype of SCN4A mutations.
ABBREVIATIONS
Skeletal muscle voltage-gated sodium channel alpha subunit 4;
Severe neonatal episodic laryngospasm;
Nicotinamide adenine dinucleotide;
Succinate dehydrogenase;
Cytochrome oxidase;
Adenosine triphosphatase;
Compound muscle action potential;
Dystrophia myotonica-protein kinase;
Zinc finger protein 9;
Heparan sulfate proteoglycan 2;
The single nucleotide polymorphism database;
The exome aggregation consortium;
Polymorphism phenotyping version 2;
Congenital muscular dystrophy;
Calmodulin-dependent protein kinase II;
Multicopy suppressor of gsp1 mutants;
Fibroblast growth factors;
Protein tyrosine phosphatase non-receptor type 3;
Synapse-associated protein 97
BACKGROUND
The skeletal muscle voltage-gated sodium channel alpha subunit 4 gene (SCN4A) is located on chromosome 17q23-25. Classically, disease-causing SCN4A mutations are dominant and cause a group of disorders called skeletal muscle channelopathies. These channelopathies are classified by the presence of episodic weakness (periodic paralysis) or muscle stiffness (myotonia); myotonia can be further subcategorized by the severity of symptoms and aggravating/alleviating factors into paramyotonia congenita, potassium-aggravated myotonia or other myotonia [1]. Although myotonia is also seen in the myotonic dystrophies, those patients develop progressive weakness and have dystrophic muscle biopsies. With the advancement of genetic testing, the phenotypic spectrum of SCN4A mutations has been expanding and now is quite broad including symptoms of exercise- or cold-induced muscle cramps, stiffness and myalgias alone [2] or neonatal stridor/severe neonatal episodic laryngospasm (SNEL) [3, 4] to congenital myopathy [5–7] or congenital myasthenic syndromes [8–10]. Congenital myopathies and myasthenic syndromes are a result of recessive mutations leading to membrane hypoexcitability, while dominant mutations lead to membrane hyperexcitability or inexcitability [11–13].
In this case study, we identified a novel de novo heterozygous SCN4A mutation associated with a complex phenotype of congenital myopathy, myotonia and congenital anomalies (Arnold-Chiari malformation, bilateral hip dysplasia, and severe scoliosis) with both classic and unique myopathic features on muscle biopsy.
CASE PRESENTATION
A 27-year-old woman presented for evaluation of a long history of episodic muscle stiffness. She was born full term to non-consanguineous parents with a prenatal course complicated by polyhydramnios and fetal hypokinesia. Multiple congenital anomalies were noted at birth including Arnold-Chiari malformation with hydrocephalus, bilateral hip dysplasia, and severe scoliosis. As an infant, she had poor sucking and feeding resulting in failure to thrive and hospitalization for intravenous nutrition support. She was floppy and not stiff after birth, according to her mother, nor did her mother recall any issues with her breathing. She did not experience any recurrent respiratory infections or need to be hospitalized for any respiratory infections as an infant or young child. Her early development was complicated by shunt placement for hydrocephalus and spinal fusion for her scoliosis which likely contributed, in part, to her delayed motor milestones. No genetic testing was pursued at that time. She first noticed stiffness at 11 years of age that increased when she walked, causing her to feel off balance and she had a tendency to fall. These episodes were not worse in the cold and did not worsen with infections or periods of fasting. She does note that her symptoms would lessen if she “warmed up”. She also had painful cramps of her lower abdomen which could occur during menses or urination. She was evaluated by gynecologist several times but no gynecology etiology was detected. She denied any episodes of paralysis or fluctuations in her weakness throughout the day. Her parents had no complaints of muscle stiffness or weakness.
On physical exam, she was of short stature with scoliosis and myopathic facial characteristics (an elongated face, blepharophimotic eyelids, low set ears and high arched palate). She did not have ptosis or any limitation of her extraocular movements. She had marked muscular hypertrophy with well-developed biceps, quadriceps, and brachioradialis muscles (Fig. 1). Eyelid, grip, and percussion myotonia were present. Her muscles strength was weak symmetrically and graded in the 4 range using Medical Research Council scale in the deltoid, biceps, and hip flexors. Although she had mild contractures in the ankles, she could walk on her toes and heels. Deep tendon reflexes, sensory and cerebellar examinations were normal. Laboratory investigations included a normal serum creatine kinase and electrolytes. Cardiac ECHO revealed mitral valve prolapse.
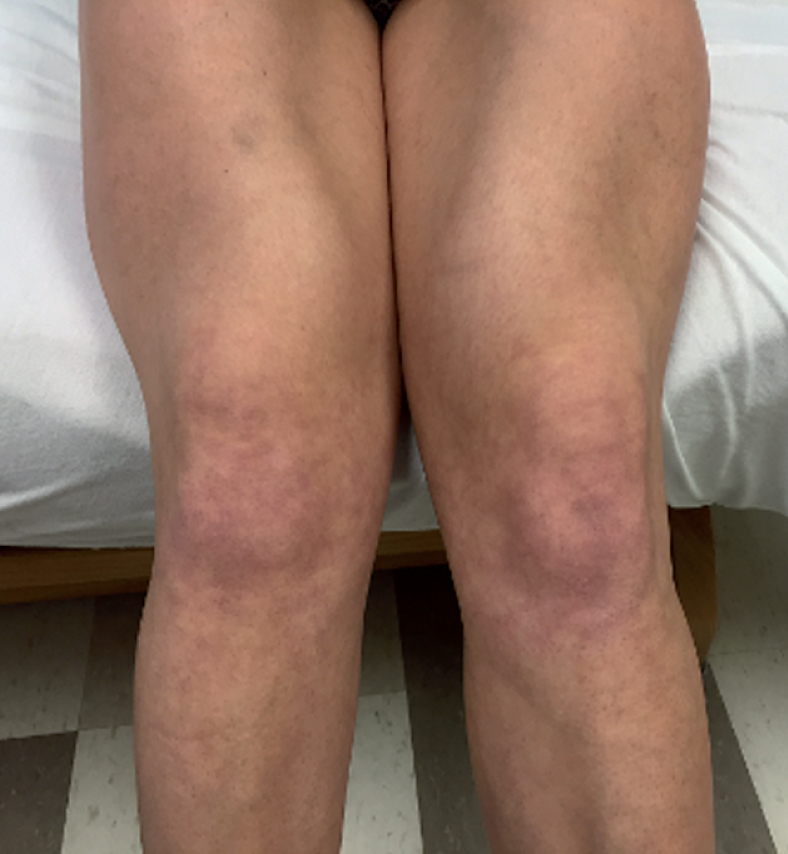
Marked quadriceps hypertrophy showing prominence of the rectus femoris with the patient in a sitting position extending her legs. The patient provided signed consent to allow for publication of this image.
Electromyography was conducted at age 33 years and revealed normal nerve conduction studies with extensive myotonic discharges and myopathic motor units in all muscles studied. Short exercise testing showed immediate post-exercise increment in compound muscle action potential (CMAP) amplitude of 22.8% at room temperature, no significant CMAP amplitude change after cooling, and decrement in CMAP amplitude of 36.3% after rewarming.
A left biceps muscle biopsy at age 27 years revealed a marked increase in variability of fiber size, numerous fibers with single or multiple internal nuclei, and rare scattered early necrotic fibers. Ragged-red fibers and tubular aggregation were not seen. The small fibers were round and displayed a lobulated pattern of oxidative enzyme distribution (NADH, SDH, and COX) while numerous large fibers were devoid of enzyme activity in central areas indicating redistribution of mitochondria (Fig. 2a–c). In the myofibrillar ATPase (pH 9.4, 4.2 and 4.6) stains, an overwhelming number of fibers were of type I histochemical fiber type and type I fiber hypotrophy was prominent and are common features seen in congenital myopathies (Fig. 2d).
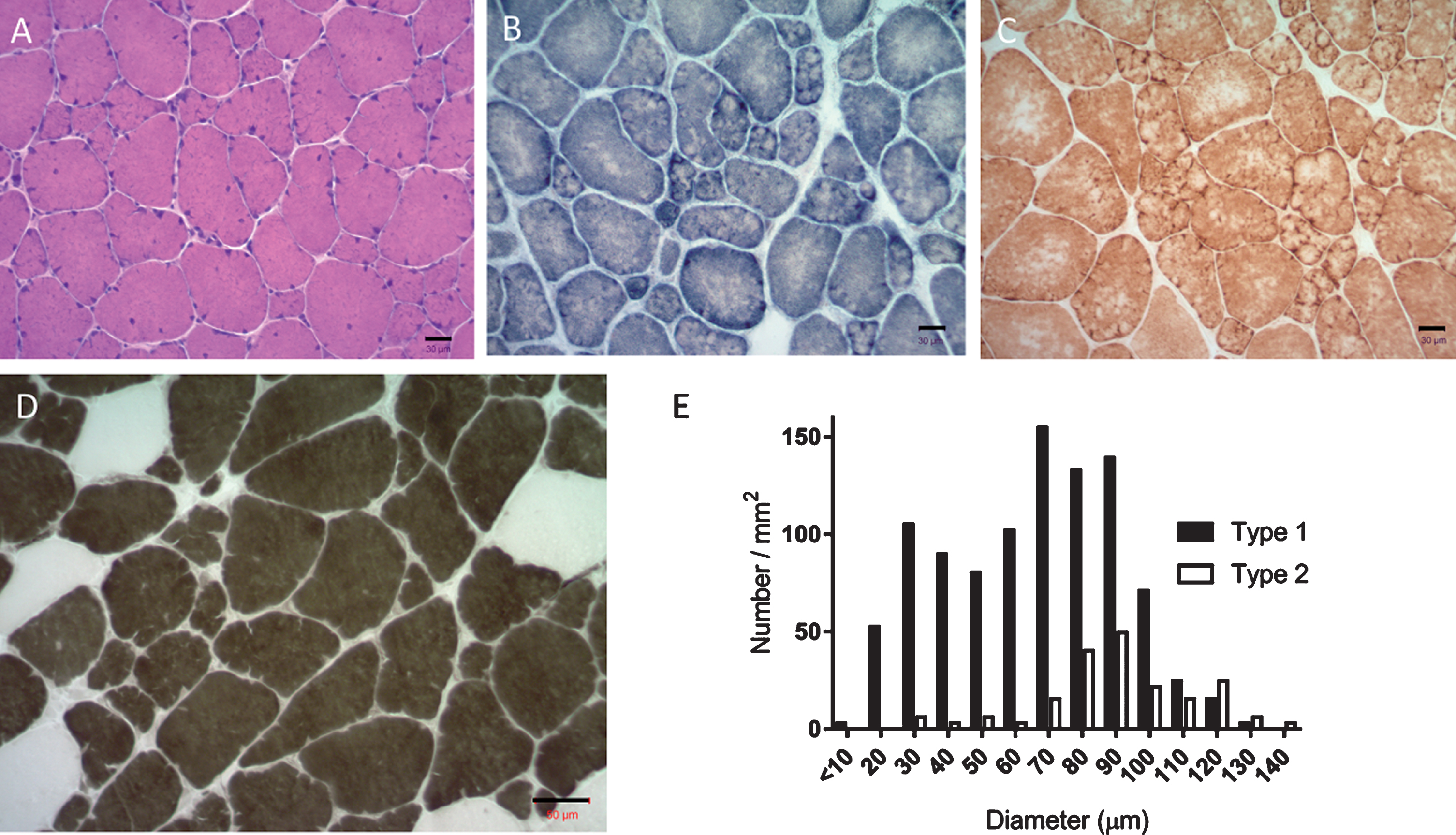
(a) H&E stained section showing muscle fiber size variability, fibers with internal nuclei and cytoarchitectural changes. (b & c) SDH and COX-reacted sections delineate the lobulated pattern of architectural change predominantly in small and medium fibers. Central areas of enzyme attenuation are noted in large fibers indicating mitochondrial redistribution. (d) Myofibrillar ATPase at pH 4.2 shows type 1 fiber predominance and type 1 hypotrophy. (e) Muscle fiber type-specific diameter distribution bar histogram. Fiber type is represented as #/mm2 for each diameter size. Three randomly photographed muscle biopsy images at 10X magnification were analyzed to determine fiber type count. A total of 315 type 1 fibers and 63 type 2 fibers were identified. Bar = 30 μm (a, b, c). Bar = 50 μm (d).
Further analysis including quantitative studies of the muscle biopsy histochemical fiber type distribution revealed type 1 fiber predominance, corresponding to 83% of total fibers counted (Fig. 2e). Type 1 fibers were 27% smaller than type 2 fibers with a mean fiber diameter of 65.76±1.43 in type 1 fibers and 90.54±2.84 in type 2 fibers (mean±SEM, p < 0.0001). The vast majority of small fibers were type 1 and showed a bimodal fiber size distribution: one subgroup composed of type 1 small fibers with diameters < 10–30 μm and a subgroup composed of larger fibers showing a peak diameter 70–90 μm. An increase in number of hypertrophic fibers with diameters over 100 μm was present in both type 1 and type 2 fibers.
Given the presence of myotonia with dystrophic muscle and myopathic facial features, genetic testing of DMPK, ZNF9, and HSPG2, which are associated with myotonic dystrophy type I (OMIM 605377), myotonic dystrophy type II (OMIM 116955) and Schwartz-Jampel syndrome (OMIM 142461), respectively, were done in sequential order with no pathologic mutations found. Subsequently, direct sequencing of the SCN4A gene revealed a heterozygous nonsynonymous variant in coding exon 14, c.2386 C>G, p.Leu796Val, which is located within the sixth transmembrane segment in the second domain of Nav1.4 channels (Fig. 3). The Leucine amino acid in position 796 is highly conserved across all species, and this variant was neither found in dbSNP [14], ExAC browser [15], as well as in 400 control samples. Using in silico analysis by PolyPhen-2 [16], SIFT [17] and Mutationtaster [18], the variant was predicted as probably damaging (score 0.999), damaging (score 100%) and disease-causing, respectively. Subsequent parental sequencing of the SCN4A gene in her mother and father did not reveal this variant and further genetic testing in the patient via the Invitae Comprehensive Myopathy panel was negative. After receiving a definitive diagnosis, the patient was started on carbamazepine and reported improvement in her symptoms. Her symptoms did not fully resolve, but she was not interested in switching to another agent, such as mexiletine, to try to ameliorate her symptoms further.
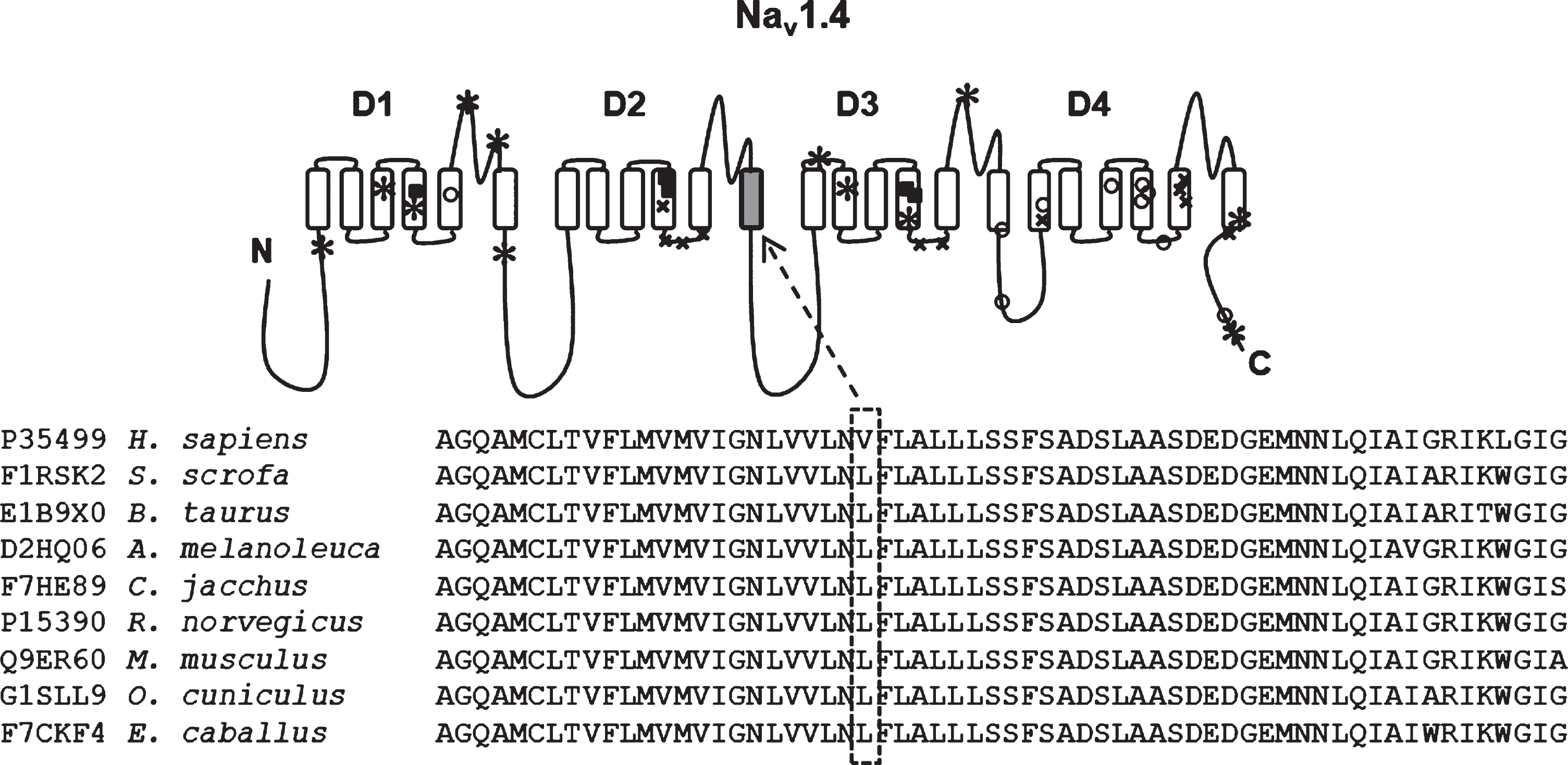
The sequences of the Nav1.4 protein from a range of divergent species, compared with multiple sequence alignment tool. c.2386C>G nucleotide change resulting p.Leu796Val substitution is located within the sixth transmembrane segment in the second domain of Nav1.4 channel. The mutated region of leucine amino acid at position 796 (highlighted with dash box) is highly conserved. Previously reported mutations are denoted in the following symbols to highlight phenotypes: *congenital myopathy; ■hypokalemic periodic paralysis; ×hyperkalemic periodic paralysis; paramyotonia congenita.
DISCUSSION
Here we report a novel, de novo, heterozygous mutation c.2386 C>G; p.Leu796Val in the SCN4A gene associated with a complex phenotype of congenital myopathy, myotonia, Arnold-Chiari malformation, scoliosis, and hip dysplasia which expands the known phenotype of SCN4A mutations. This p.Leu796Val mutation in SCN4A is highly conserved across all different species, and this variant was predicted as disease-causing, and thus, we believe this mutation has a high probability of causing the reported patient’s gain-of-function and loss-of-function phenotypic characteristics.
Recessive SCN4A mutations causing a loss-of-function or partial loss-of-function are becoming well known causes of congenital myopathy. Eleven patients were reported in 2016 with recessive (homozygous or compound heterozygous) SCN4A mutations who had severe congenital hypotonia, intrauterine contractures, significant bulbar dysfunction and spinal deformities; seven of whom died prior to, or shortly after, birth [5]. Muscle biopsies of the four surviving patients showed fiber size variability and fibrofatty tissue without specific structural abnormalities. In 2017, an additional three patients were reported with recessive (compound heterozygous) SCN4A mutations who had polyhydramnios, severe hypotonia, cryptorchidism, myopathic facies, severe swallowing dysfunction requiring tube feeding and delayed the acquisition of motor skills. Again, all patients had non-specific findings on muscle biopsy [6]. In the case presented here, the patient clearly has features of classic congenital myopathy (polyhydramnios, requiring nutritional support due to poor suck/feeding, high arched palate with an elongated face) but in the setting of a novel, dominant SCN4A mutation, rather than a recessive mutation.
Dysmorphic and myopathic features are not typically seen in the dominant skeletal sodium channelopathies, though rare cases have been reported. Yoshinaga H et al. reported a boy who carried a heterozygous c.2077A>C (p.Ile693Leu) mutation in SCN4A with overlapping features of myotonia and periodic paralysis and features resembling Schwartz-Jampel syndrome (low-set ears, epicanthic folds, upturned nose, a long philtrum, puckered lips, short neck, hypertrophic thighs, atrophic shoulder girdle muscles, pigeon breast, and joint contracture of the elbow) [19]. Fusco C et al. also reported a 4-year-old girl who carried a heterozygous c.3539A>T (p.Asn1180Ile) mutation in SCN4A gene which presented with diffuse stiffness most predominant in the lower limbs and abdomen in conjunction with facial and skeletal dysmorphisms (high forehead, down-slanting palpebral fissures, short neck, and high arched palate, clinodactyly, bilateral clubfoot, pes cavus and hip dislocation) which were presumed to be caused by fetal hypokinesia [20] It should be considered that myopathic facies, scoliosis and hip dysplasia, which are now reported twice (Fusco et al and here), are attributable to SCN4A mutations. Although there have been no reports of Arnold-Chiari malformations associated with SCN4A mutations to date, there has been one report of synostosis of the sagittal and metopic sutures reported previously [7]. In considering the known associations between craniofacial malformations, craniosynostosis, and Arnold-Chiari malformations [21–23], it is possible that these congenital malformations may also represent a spectrum of features resulting from SCN4A mutations.
Muscle histology in SCN4A mutations can be normal to non-specific [2] in milder phenotypes and non-specific to myopathic in more severe/recessive phenotypes [5]. Recently Gonorazky et al reported two patients with a compound heterozygous SCN4A mutation and muscle biopsy features of a central core-like area of disorganization surrounded by a crown of nuclei which they propose to call “corona fibers” in two siblings with congenital myopathy [7]. Here we note features of marked fiber size variability, type I fiber predominance and type I fiber hypotrophy and hypertrophy with minimal scattered necrosis and regeneration that are typical of a congenital myopathy in the setting of a dominant mutation. The additional finding of an overwhelming number of type I small and medium fibers, having a lobulated structural pattern was quite interest and is thought to be associated with impaired regeneration as we have shown in the calpain 3 knockout mouse model [24]. Lobulated type I fibers can be seen in other conditions, rather commonly in calpainopathy, Ullrich CMD, or Bethlem myopathy.
Molecular genetic analysis revealed a novel de novo heterozygous c.2386 C>G missense mutation in the SCN4A gene causing p.Leu796Val amino acid change that is likely to be critical since the leucine in this particular position is highly conserved across all species and has not been reported to our knowledge. Most heterozygous/dominant SCN4A mutations result in a periodic paralysis or myotonia phenotype due to missense gain-of-function mutations which can alter channel gating, cause partial disruption of inactivation, or enhanced activation leading to propagation of the action potential [11–13]. In mild cases, this leads to myalgias, myotonia or paramyotonia and in severe cases, it leads to periodic paralysis. As noted above, in contrast, homozygous or compound heterozygous SCN4A mutations result in a congenital myopathy or congenital myasthenic syndrome phenotype due to a loss-of-function (impaired or absent action potentials) [11–13]. Notably, only SCN4A mutations that result in hyperkalemic periodic paralysis and cause enhanced inactivation via gating pore leakage currents can lead to a mixed phenotype of gain- (myotonia) and loss-of-function (paralysis). To date, we are unaware of any other heterozygous SCN4A mutation causing such a disparate mixed phenotype of congenital myopathy and myotonia.
We are proposing that this heterozygous c.2386 C>G missense mutation is causative. It is well known that SCN4A mutations can have variable penetrance even within families [19, 25]. Furthermore, the recessive p.Arg1135Cys mutation leads to hypokalemic periodic paralysis but when a compound heterozygous mutation in conjunction with p.Arg104His, it leads to a classic congenital myopathy [5, 26]. Thus, it is may be possible for the heterozygous mutation described here to cause such a unique phenotype. Though beyond the scope of this report, it will be important to do functional studies of this mutation in cells to determine/prove how it can cause both a gain-of-function and loss-of-function phenotype. Finally, Loussouarn et al note that several proteins (dynamitin, 14-3-3, CaMKII, MOG-1, calmodulin, FGF, PTPH1/PTPN3 and SAP97) that interact with Nav1.5 on a site conserved in Nav1.4 may also play a role in altering skeletal sodium channel function [25]. These proteins are known to alter Nav1.5 function (cardiac muscle) and may likely alter skeletal muscle as well. Although a congenital myopathy panel was negative for any further variants in the patient reported here, further investigation for additional modifiers in known SCN4A patients may explain the growing phenotypic variability.
In conclusion, we report a novel heterozygous mutation in SCN4A associated with a complex phenotype and distinct muscle biopsy features. Here we report a SCN4A mutation resulting in a mixed gain-of-function (myotonia)/loss-of-function (congenital myopathy) phenotype and the first report of a dominant SCN4A mutation causing a congenital myopathy. This study adds to the expanding the knowledge of SCN4A mutations and their varied phenotypes.
CONSENT FOR PUBLICATION
The patient reported here provided written informed consent to publish their data. A copy of the written consent has been made available for review by the editor of this journal.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
AUTHORS’ CONTRIBUTIONS
All authors confirmed that they have contributed to this paper. ZS performed specialized medical examinations of the patient, coordinated the study, evaluated the histopathological, molecular and medical data, and approved the final draft of the manuscript. JA and MW coordinated the study, evaluated the molecular and medical data and wrote the manuscript. CP and DB evaluated the histopathological data. All authors read and approved the final manuscript.
Footnotes
ACKNOWLEDGMENTS
The authors wish to acknowledge Sarah Lewis and Kristin Battles at the Neuromuscular Pathology Laboratory, Center for Gene Therapy, Nationwide Children’s Hospital for their excellent technical support.