
Abstract
TRIM63 mutations have been described as a potential cause for cardiac and skeletal myopathy in only one family so far. We describe a new patient carrying the same homozygous TRIM63 nonsense mutation c.739 C>T p.Q247X, that was originally reported in two members of a Spanish family manifesting cardiac hypertrophy. One of these original patients also had an additional heterozygous mutation in TRIM54 and a much more severe phenotype also involving skeletal muscles, and a digenic inheritance was therefore suggested. Our case report confirms the role of TRIM63 as a new cardiac myopathy gene, although it is unclear whether the homozygous p.Q247X mutation alone is sufficient to cause an additional skeletal myopathy.
Keywords
ABBREVIATIONS
Tripartite motif 63/ Muscle Ring-Finger1 (TRIM63/MuRF1)
Tripartite motif 54
transient ischemic attack
Tripartite motif 63/ Muscle Ring-Finger1 (TRIM63/MuRF1) protein is an important regulator of muscle atrophy and is widely expressed in both skeletal and cardiac muscle [1]. In a previous publication, heterozygous mutations in the TRIM63 gene were more commonly detected in a cohort of hypertrophic cardiomyopathy patients than in controls (4.4% vs 1.1%) [2]. Furthermore, mice lacking MuRF1 protein were shown to develop exaggerated cardiac hypertrophy in response to both physiological and pathological myocardial strain compared with wild-type mice [3].
A digenic inheritance was proposed in a Spanish protein aggregate myopathy (PAM) patient carrying both a homozygous nonsense mutation null allele c.739 C>T p.Q247X in TRIM63 combined with a heterozygous missense mutation in the homologous tripartite motif 54 (TRIM54 gene) [4]. We report a patient with mild skeletal myopathy and cardiac hypertrophy carrying the previously reported homozygous p.Q247X mutation.
CASE REPORT
The patient was initially investigated for a transient ischemic attack (TIA) at age 56 years. On routine etiological work-up because of the TIA, she was noted to have an elevated creatine kinase at 700 IU/l (normal <210 IU/l) as well as cardiac hypertrophy on ultrasound. On cardiac MRI, the maximal left ventricular wall thickness was 14 mm (normal values for women are less than 11–12mm). Her blood pressure has always been normal without medication (<120/80 mmHg). Nerve conduction studies were normal and EMG showed mild myopathic changes in proximal upper and lower limb muscles. Muscle biopsy from vastus lateralis showed mild myopathic changes. Lower limb muscle MRI displayed mild diffuse fatty infiltration in the right gluteus minimus, both vastus lateralis and semimembranosus muscles and the right medial gastrocnemius.
At age 65, she was still fully ambulatory without aids, but reported mild fatigability of upper limb muscles that did not interfere with daily activities. A thorough neurological examination including muscle strength testing at age 65 was normal. Her cardiac function has remained stable over a follow-up of 9 years based on repeated echocardiographic examinations and no obvious disease progression was noted with a muscle MRI performed 10 years after the first one (Fig. 1). A gastrocnemius medialis muscle biopsy showed a few necrotic fibers, fiber size variability and increased number of internal nuclei. A striking finding was the presence of multiple internalized capillaries inside the muscle fibers in 5–10% of the fibers (Fig. 2). There was no family history suggestive of neuromuscular or cardiac disorders. The patient’s leukocyte-derived DNA was examined with our next generation sequencing panel (MyoCap version 3) covering 265 known or suspected skeletal- and cardiomyopathy-related genes [5]. The MyoCap data was also assessed for possible deletions of duplications using our previously reported pipeline [6]. The patient was shown to carry the same homozygous nonsense mutation p.Q247X previously reported in association with hypertrophic cardiomyopathy and digenic protein aggregate myopathy [4]. No mutations or copy number variations were found in the TRIM54 gene.
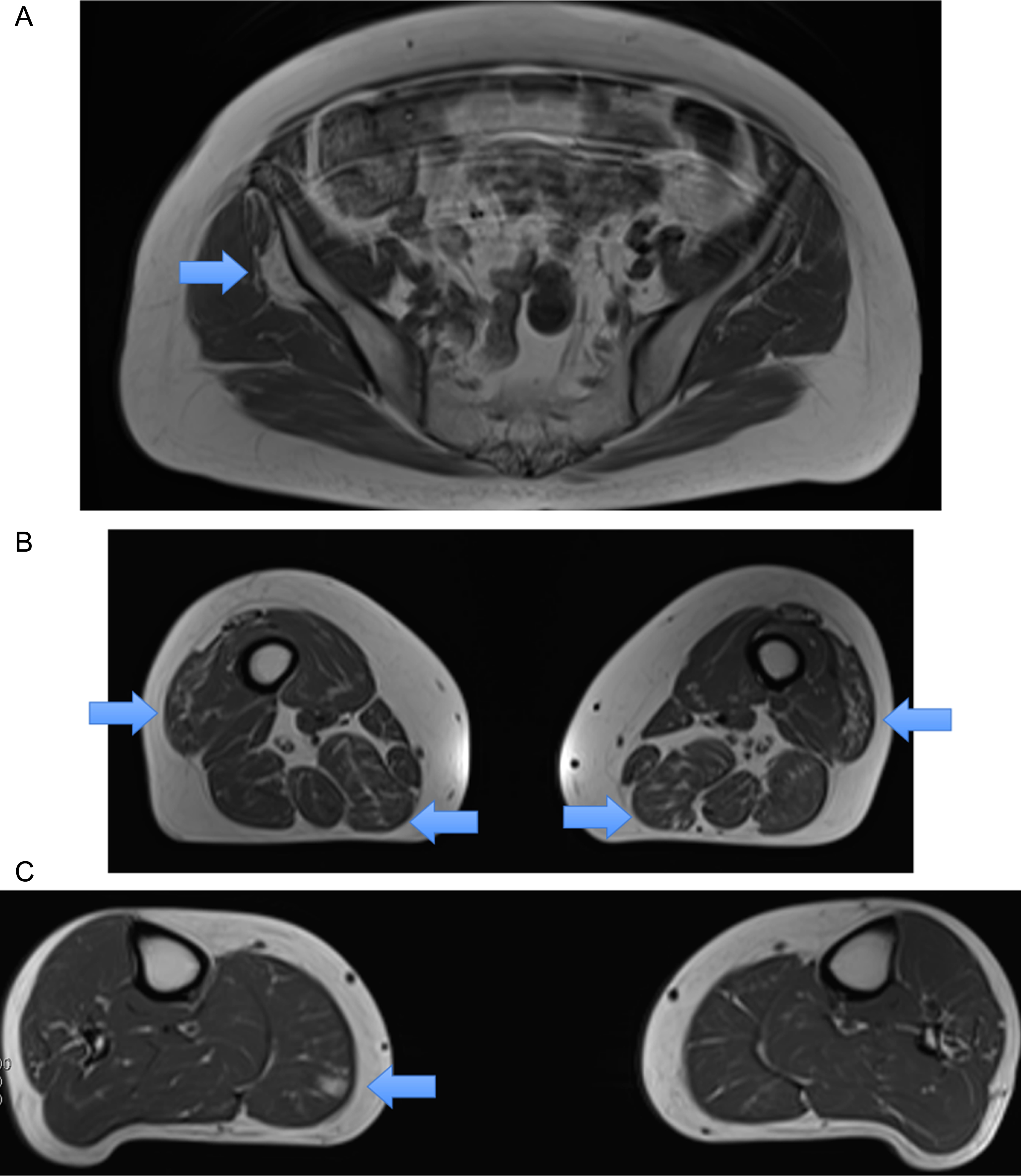
Lower limb muscle MRI displayed mild diffuse fatty infiltration in the right gluteus minimus (A, arrow), both vastus lateralis and semimembranosus muscles (B, arrows) and the right medial gastrocnemius (C, arrow).
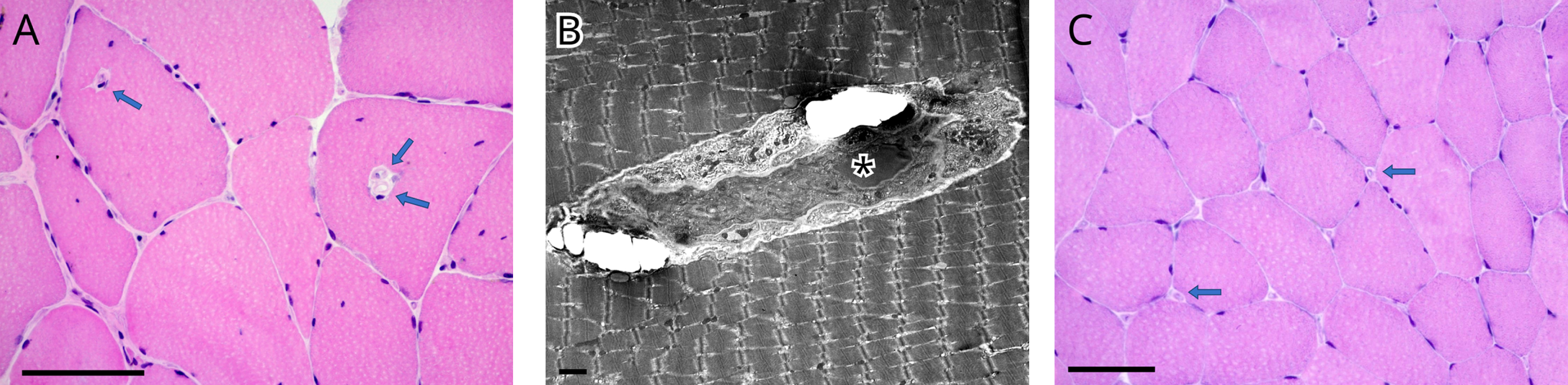
Internalized capillaries were seen in the medial gastrocnemius in about 5–10% of muscle fibers (A, B) while normally they are located around muscle fibers (C, control muscle from another patient showing the normal location of capillaries). Fiber size variation and internal nuclei were also detected (A). B = electron micrograph. A, C = Haematoxylin and eosin stain. Scale bar = 00 micrometer (A, C), scale bar = micrometer (B).
All procedures described in this report were performed in accord with the ethical standards of the Helsinki Declaration of 1975.
DISCUSSION
We report a patient carrying the same homozygous TRIM63 nonsense mutation p.Q247X, which in a recent article had been identified in two members of a Spanish family (identified as III-3 and III-4), both of whom had cardiac hypertrophy. Patient III-4 with the homozygous TRIM63 p.Q274X mutation in combination with a heterozygous TRIM54 variant had a severe adult-onset protein aggregate myopathy. This is in contrast with the paucisymptomatic phenotype observed in our patient, who did not show protein aggregates on muscle biopsy. In the original Spanish family, a female relative (III-3) carrying only the homozygous TRIM63 mutation was found to have cardiac hypertrophy but was not examined in detail for a possible mild skeletal myopathy. Our report is therefore compatible with the suggested digenic mechanism modifying the phenotype in the Spanish family and leading to a more severe phenotype with the combined TRIM63/TRIM54-mutations than with the homozygous TRIM63-mutation alone.
In conclusion, we suggest that physicians should also query for symptoms and signs of skeletal muscle disease while evaluating cardiomyopathy patients carrying the homozygous TRIM63 mutation Q274X. However, even though cardiac hypertrophy has been detected in all three reported patients reported so far, it is unclear whether this homozygous mutation alone is sufficient to cause an additional skeletal myopathy or whether this requires the presence of some additional environmental or genetic factors.