
Abstract
Autosomal dominant centronuclear myopathy (CNM) caused by mutations in the gene coding for amphiphysin-2 (BIN1) typically presents in adulthood with progressive muscle weakness. We report a Dutch family with AD CNM due to a novel BIN1 mutation (c.53T>A (p.Val18Glu)), strongly impairing the membrane tubulation activity of amphiphysin-2. The main features were mild proximal weakness with pronounced myalgia, exercise intolerance and large muscle mass, with a childhood onset in the youngest generation and mild cognitive features. This suggests BIN1 mutations should be considered in patients with isolated exercise intolerance and myalgia, even in childhood.
INTRODUCTION
Centronuclear myopathies (CNM) are a group of congenital myopathies characterized by type I fiber predominance and increase of central nuclei [1–6]. Recent research has identified adult-onset autosomal dominant (AD) CNM caused by mutations in the gene coding for amphiphysin-2 (BIN1) [7]. The mutations either affect the N-terminal amphipathic helix of amphiphysin-2, or involve a loss of the stop codon and the inclusion of 52 supernumerary amino acids [7, 8]. Typically, these patients presented in adulthood with progressive muscle weakness. Recently, proximal weakness with myalgia and CK elevation was reported in an Italian family with a mutation in the N-terminal amphipathic helix (p.Ala36Glu) [8].
We here report six patients belonging to a Dutch family with AD CNM due to a novel mutation in BIN1 c.53T>A (p.Val18Glu), strongly impairing the tubulation properties of amphiphysin-2. The main clinical features were mild proximal weakness with pronounced myalgia, exercise intolerance and large muscle mass, with a childhood onset in the youngest generation and mild cognitive features.
CASE REPORT
Patient 2.1
The proband is a 76-year old woman. She has ten siblings, two of which were also diagnosed with AD CNM. She has had short Achilles tendons since childhood and had never been very good at sports. In her fifties, she sought medical attention for chronic lower back pain. Gradually, the muscle strength in her upper and lower limbs decreased, and walking and stair climbing became increasingly difficult. At the age of 75, she was diagnosed with parkinsonism and dementia.
Clinical examination (age 67) revealed calf and biceps hypertrophy, scoliosis, and mild bilateral scapula winging (Fig. 1 and Table 1). Muscle weakness was more pronounced distally than proximally, and flexors were more affected than extensors. She had a waggling gait, bilateral drop foot, and was unable to walk on her heels, caused by Achilles tendons contractures. CK level was normal (64 u/l). Cardiac and respiratory assessments were normal. EMG showed generalized myopathy.
CT of the muscles (age 65) showed fatty replacement of the muscles of the lower back and hamstring muscles with sparing of the biceps femoris. Also fatty replacement was present in the tensor fascia lata, adductor magnus, and in the muscles of the posterior compartment of the lower legs. Muscle biopsy of the quadriceps revealed an increase of internal and central nuclei with occasional nuclear clustering and fiber splicing. Mutations of MTM1, DNM2 and RYR1 were excluded before.
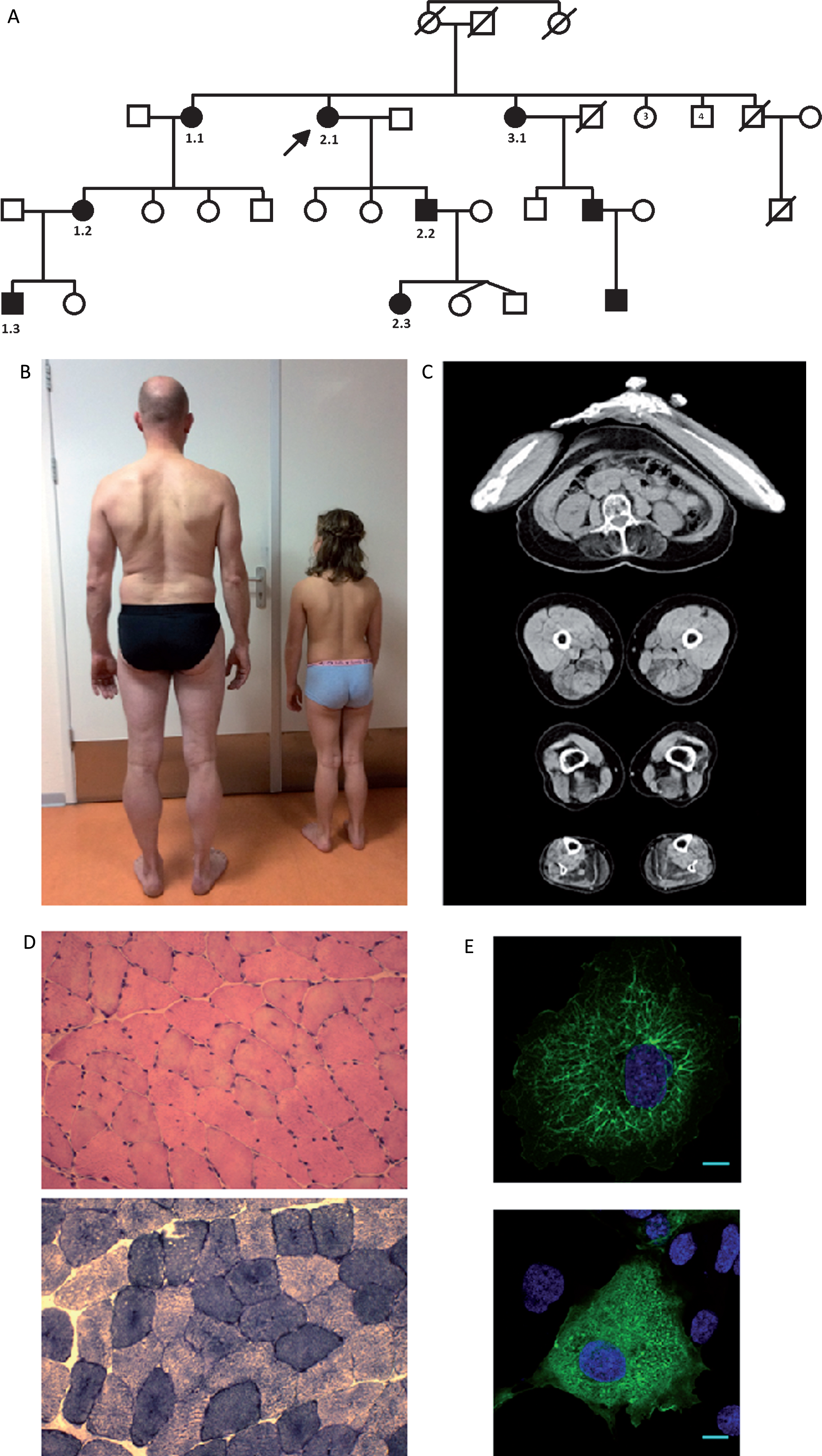
Clinical, histological and functional findings in an AD CNM family with a novel BIN1 mutation. (A) Pedigree of family with AD CNM and BIN1 mutation. Marked are the affected patients. (B) Muscular stature with calf hypertrophy in patient 2.2 and 2.3. (C) CT scan of index patient (2.1) demonstrating bilateral fatty replacement of the muscles of the lower back, hamstring muscles with sparing of the biceps femoris. Also fatty replacement is present in the tensor fascia lata and adductor magnus, and in the muscles of the posterior compartment of the lower legs. (D) Hematoxilin & eosin staining of the quadriceps muscle revealing increased internalization and centralization of myonuclei. There was a slight increased percentage of type I fibers. (E) Exogenous expression of GFP-BIN1-WT in COS-1 cells induces membrane tubulation (upper figure is wild type). In contrast, cells transfected with the GFP-BIN1 construct harboring the p.Val18Glu mutation do not display membrane tubules (lower figure). Scale bar corresponds to 10 μM.
This table shows the clinical features and findings of ancillary investigations in the three branches of this family. The index case is patient 2.1. In braches 1 and 3 of this family, age of onset was lower in subsequent generations (family 1 based on investigation; family 3 based on family history)
N.d. = not determined. ADHD: attention deficit and hyperactivity disorder.
Follow-up at the age of 75 years showed features of a hypokinetic rigid syndrome: mild bradyphrenia, a forward flexed posture, an asymmetric rest tremor and hypokinesia, with dysarthria, hypophonia and drooling. MR of the brain showed aspecific white matter changes, and a DAT scan showed a signal compatible with nigrostriatal dopaminergic degeneration.
Patient 2.2
The son of patient 2.1 was investigated at the age of 35 years. Motor development was normal. His medical history showed meningitis (age of 11 years) with full recovery. He had adjusted footwear for Achilles tendons contractures since childhood. During follow-up, he had developed painful muscle stiffness predominantly in the upper limbs.
Physical examination (age 45) showed calf hypertrophy, Achilles tendon contractures, and paravertebral muscle atrophy. Mild weakness of the rhomboid muscles (MRC4) was observed. No muscle weakness of the forearms, hands, or lower limbs was detected. EMG showed myopathic changes. The quadriceps muscle biopsy revealed increased internalization and centralization of myonuclei. There was a slightly increased percentage of type I fiber. Muscle MRI was normal. Neuropsychological testing showed no abnormalities.
Patient 2.3
The daughter of patient 2.2 first presented at the age of three years. Pregnancy and delivery were uneventful. She had an asymptomatic atrial septal defect. Motor milestones were normal, with walking at 16 months. She often complained about tight calves and sore legs, in particular after exercise. A year later painful nighttime muscle cramps in her legs developed, for which ibuprofen and transcutaneous electrical nerve stimulation were prescribed with limited effect. She had difficulties walking stairs and running. She had surgery for strabismus at the age of six. At school, she had difficulties concentrating and processing information.
Physical examination at the age of 8 years showed calf hypertrophy, Achilles tendons contractures, short hamstring muscles, and bilateral pes planovalgus. She had mild bilateral winging scapula, mild lumbal lordosis, and atrophy of the paravertebral thoracic muscles. Muscle weakness was observed axial and proximal in the upper and lower limbs. She had an atrial septal defect but no other cardiac involvement. Respiratory assessments was normal. CK level was elevated 3-4 fold normal value (567 U/L). Muscle biopsy of the quadriceps muscle revealed enlarged mitochondria with no signs of myopathy. Psychological assessment (WPPSI-III-NL) showed reduced intelligence quotient (IQ) of 77 (borderline) [11]. The intelligence profile was disharmonic, resulting in a significantly higher verbal IQ (VIQ 89) than the performance IQ (PIQ 67). The processing speed quotient was just below average (PSQ 85). The general language composite was average (GLC 91).
Patients from two other branches of the family presented similar symptoms, including the observation that the younger generation developed symptoms earlier.
Molecular genetics
Genomic DNA was prepared from peripheral blood by routine procedures and sequenced for all coding exons and intron/exon boundaries of BIN1 by bi-directional Sanger sequencing [6]. The mutation was numbered according to GenBank NM_139343.2 and NP_647593.1. BIN1 sequencing revealed a heterozygous c.53T>A (p.Val18Glu) missense mutation in exon 1 in all affected family members.
Functional assessment
In order to assess the functional impact of the BIN1 c.53T>A (p. Val18Glu) mutation, we generated and transfected wild type and mutant GFP-BIN1 constructs into COS-1 cells. In transfected cells, exogenously expressed wild type BIN1 promotes membrane tubulation in the vast majority of the transfected cells. In contrast, the BIN1 construct harboring the p.Val18Glu mutation did not induce tubulation in any cell. This is in accordance with previously reported heterozygous BIN1 mutations also affecting the N-terminal amphipathic helix as p.Lys21del, p.Arg24Cys, or p.Ala36Glu, and represents a strong functional proof for the pathogenicity of the mutation in BIN1.
DISCUSSION
This study shows a three-generation family with AD CNM due to a novel BIN1 mutation. The p.Val18Glu mutation detected in this family affects a conserved residue of amphiphysin 2, and is predicted to strongly impact on the hydrophobic face of the N-terminal amphipathic helix, which plays an essential role in membrane curvature induction [9, 10]. Accordingly, exogenously expressed amphiphysin 2 harboring the p.Val18Glu mutations was unable to induce membrane tubulation.
This report includes a remarkable intrafamilial variability. First, myalgia and cramps, together with exercise intolerance were the most debilitating features in the son and granddaughter of the index patient. AD CNM due to heterozygous BIN1 mutations can thus correlate either with progressive muscle weakness and/or myalgia as main signs. This confirms the report of Girabaldi et al. on a patient with mild proximal and girdle weakness and pronounced myalgia [8], and suggests BIN1 mutations should be searched in patients with isolated exercise intolerance and myalgia.
Furthermore, the third generation had an onset in childhood, whereas the first and second generation reported an onset in (young) adulthood. This might be related to the interfamilial variability but also to the closer medical follow up of the youngest patient. The previous autosomal dominant cases all had an adult onset (52 years in the proband reported by Girabaldi et al.; 22 years in the series reported by Bohm et al.) [7, 8]. Thus, this study reveals dominant BIN1 CNM can have an early presentation and can be encountered by pediatricians. Finally, four patients in this family had features of central nervous system involvement (disharmonic mental development, attention deficit, memory disturbances and parkinsonism and dementia), which has so far only been reported in the recessive cases. The central nervous system involvement might be related to the significant expression of the BIN1 isoforms containing the mutated exon 1 in brain. In short, this report has shown a new mutation in BIN1 (c.53T>A (p. Val18Glu)) with childhood onset of myalgia and cramps and hints of CNS involvement.
DISCLOSURES
Dr Kouwenberg reports no disclosures
Dr Bohm reports no disclosures
Dr Erasmus reports no disclosures
Dr van Balken reports no disclosures
Dr Vos reports no disclosures
Dr Kusters reports no disclosures
Dr Kamsteeg reports no disclosures
Dr Biancalana reports no disclosures
Dr Koch reports no disclosures
Dr Dondain reports no disclosures
Dr Laporte reports no disclosures
Dr Voermans reports no disclosures
The functional assessment was performed with support of the French Muscular Dystrophy Association (AFM-Téléthon; AFM 17088).
CONFLICTS OF INTEREST
The authors have no conflict of interest to report.
Footnotes
ACKNOWLEDGMENTS
We are grateful to the patients who participated to this study.