
Abstract
INTRODUCTION
The L25 line of transgenic mice arose by chance from a study of ocular lens development. Several lines of transgenic mice were generated by random genomic integration of a transgene encoding Sef b (an antagonist of fibroblast growth factor receptor signalling) under lens-specific transcriptional control [1]. While maintained as hemizygotes (L25+/–), all the lines showed wild-type motor behaviour. However, L25+/– × L25+/– matings yielded a subset of homozygous (L25+/+) progeny with a severe motor phenotype. Genomic sequencing, conducted only after completion of the phenotype study, revealed that the transgene had, by chance, disrupted the beta-IV spectrin gene. Western blotting confirmed reduced beta-IV spectrin protein levels in the brain. Deliberate targeting of the beta-IV spectrin gene was previously shown to hinder the clustering of voltage-gated sodium channels at Nodes of Ranvier and axon proximal segments on central neurons, thereby explaining the reported impairment of motor control[2, 3].
Here we have quantified the postnatal onset and progression of motor signs in affected L25 mice arising from L25+/–×L25+/– matings. The signs of motor impairment varied somewhat from animal to animal. The most consistent endpoint determinant was the loss of body weight crossing the predefined endpoint for euthanasia.
MATERIALS AND METHODS
This study was approved by The University of Sydney Animal Ethics Committee in compliance with the NSW Animal Research Act 1985 and the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes 8th Edition NHMRC 2013. Six breeding pairs of L25+/– mice were mated to generate a total of 148 progeny. Wild-type pups were identified by transgene PCR [1]. Mice were held in filter top cages with dimensions 17×34×l3 cm (width×length×height), with free access to water and YSF branded mouse and rat chow with a 12 hour light/dark cycle and room temperature set to 21°C. Environmental enrichment consisted of paper tissues, a plastic igloo and a 4 cm diameter cardboard tube. At weaning, each pup was ear-marked and the male and female pups were separately group-housed. Genomic PCR was used to detect the presence/absence of the transgene [4]. From weaning, phenotype observations were made weekly by EK, who remained blind to the genomic PCR results until each litter reached 8 weeks of age. Each pup was placed individually in an empty, open topped Perspex observation cage. A one-minute video clip was used to score spontaneous motor activity (number of grid squares crossed; see also Supplementary methods). The mouse was killed by intraperitoneal injection of pentobarbitone (30 mg) as soon as it displayed any one of the following pre-specified termination criteria: 1/ grade II weakness (lying prone/flaccid at rest; [5]), 2/ loss of ≥15% of its peak body weight, or 3/ unable to right itself within 10 seconds when turned on its back [6]. Immunofluorescent labeling and morphometric analysis of neuromuscular junctions was performed as previously described [7].
Genomic DNA was extracted from the ear of an affected mouse using the DNeasy Blood & Tissue Kit (QIAGEN, Valencia, CA, USA) according to manufacturer instructions. Library preparation for whole genome sequencing was conducted using the TruSeq DNA PCR Free kit (Illumina, San Diego, CA, USA) and paired-end sequencing was performed on the HiSeq X Ten instrument (Illumina, San Deigo, CA, USA). For bioinformatics analysis, raw FastQ data were aligned to the mouse reference genome mm10 using the Burrows-Wheeler Aligner v0.7.12 (BWA; [8]). To confirm the presence of the alpha AV3 sequence in the sample, unmapped mm10 reads were extracted using SAMtools v0.1.3 [9] and mapped to the alpha AV3 sequence using BWA v0.7.12 and visualised using the integrative genomics viewer (IGV; [10]. To predict the specific alpha AV3 insert position in the mouse genome, raw FastQ data was simultaneously aligned to mm10 and alpha AV3 using BWA v0.7.12. Chimeric reads (reads containing sequences from mm10 and alpha AV3) were obtained using SAMtools v0.1.3 and visualised using IGV to determine the transgene insert position.
Western blotting of SDS-PAGE gels was used to compare the levels of βIV spectrin in brain homogenates, based on previous studies (11; see Supplementary methods for full details). In brief, the Micro BCA protein assay reagent kit (Thermo Fisher Scientific) was used to measure protein concentrations of homogenised brain lysates and to ensure equal gel loading. Following SDS-PAGE, bands were transferred onto a polyvinylidene fluoride membrane, which was subsequently probed using a monoclonal antibody specific for β-IV spectrin (clone N393/76; Merck Millipore), or GAPDH (G8795; Sigma-Aldrich Corp.). Non-immune, isotype-control IgG2b (MPC-11; monoclonal, Merck Millipore) was used to exclude possible non-specific binding by the primary antibody.
RESULTS
Between 3–5 weeks postnatal, 19.5% of the L25+/–×L25+/– progeny (29/148 pups) were identified by a fine, sustained whole-body tremor, together with a hind-limb retraction response when lifted by the tail (Fig. 1A and C ). The retraction response contrasts with the reflex splaying of the hind limbs found with wild-type mice (Fig. 1B and D). These affected L25 mice, both male and female, were slower growing than wild-type littermates (Fig. 1I and J). Affected male and female L25 mice moved about much less when placed in an empty cage, compared to wild-type littermates (Fig. 1K and L). The reduced motor activity was likely explained by difficulty controlling the hind limbs during locomotion (Fig. 1E and G; c.f. wild-type littermate Fig. 1F and H; Supplementary video).
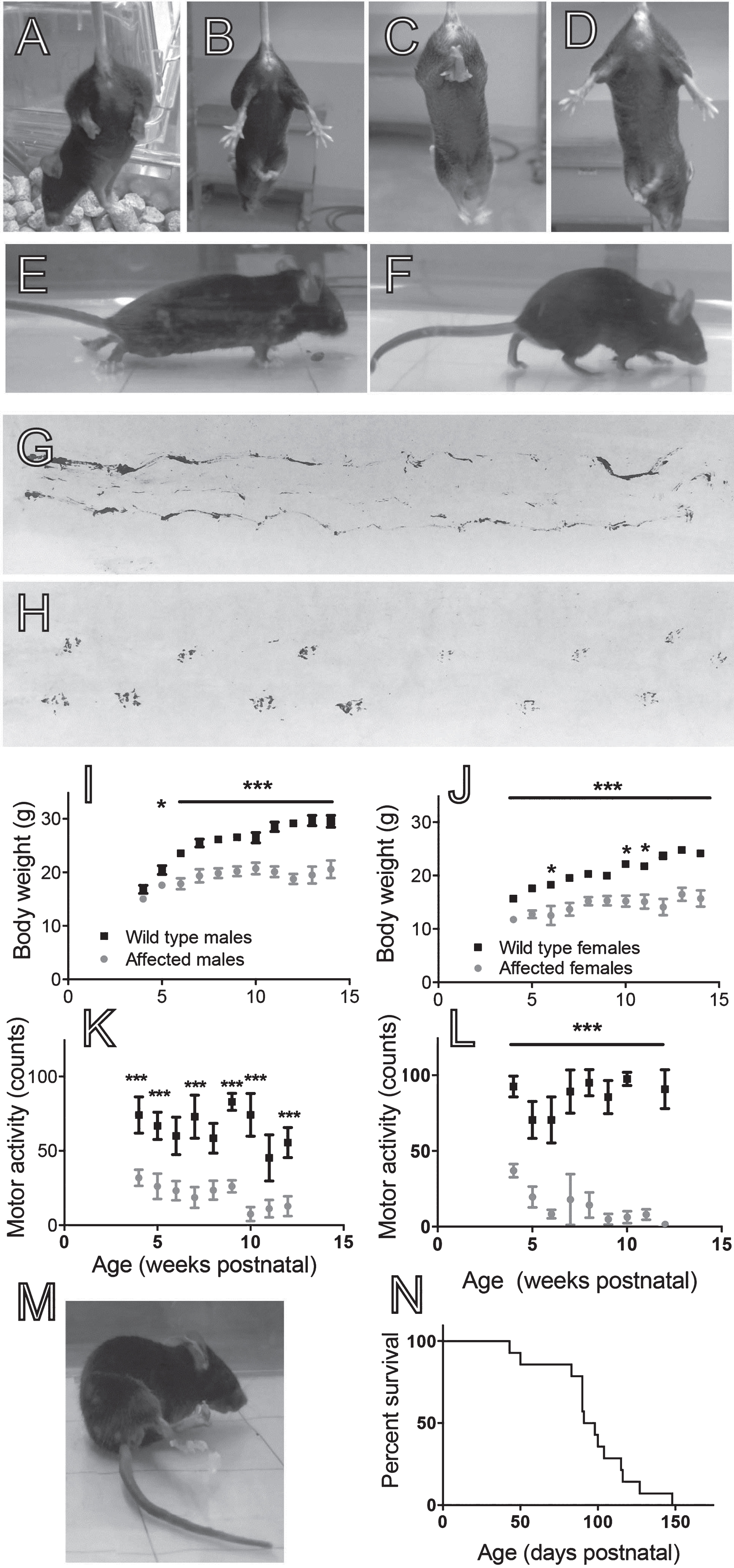
Phenotype of affected L25 transgenic mice. (A) A one-month old L25 mouse displaying mild hind limb clenching. (B) Wild-type littermate showing normal hind limb splaying. (C) Affected L25 mouse displaying severe clenching with balling of the foot at the endpoint. (D) Wild-type littermate. (E) Body posture of an affected L25 mouse at the endpoint. (F) Wild-type littermate for comparison. (G and H) Hind limb footprint records from an affected L25 mouse and wild-type littermate respectively. (I) Body weights of affected L25 males compared to wild-type controls with age. (J) Weights of affected L25 females and wild-type controls. (K) Spontaneous motor activity for affected male L25 mice and wild-type controls. (L) Motor activity for affected female L25 mice and wild-type controls. Bars represent mean±SEM with n = 9 mice in each sample. Statistical significance was determined using the Holm-Sidak method, not assuming equal standard deviations (*P < 0.05, ***P < 0.001). (M) Spastic paralysis of the hind limbs at the ethical endpoint. (N) Survival curve (ethical endpoint) for affected L25 mice (n = 14:9 males plus 5 females).
A total of 14 affected L25 mice were followed through to their ethical endpoint, which ranged between 43 and 148 days postnatal. The half-survival age for affected L25 mice was 92 days (Fig. 1N). In all cases (14/14 mice) euthanasia was dictated by the pre-specified ≥15% loss of body weight (mean 19%, range 16–23%). This weight loss usually occurred within a 1-2 week period. At the endpoint, all animals still displayed hind limb retraction when lifted by the tail. Half of the mice (including all 5 females) also displayed an intense balling of the foot during attempts at locomotion, together with either spastic paresis or spastic paralysis of the hind limbs. Most of the affected males (7/9), displayed a distinct body tremor at endpoint. Supplementary Table S1 lists endpoint motor signs for all 14 affected mice.
Neuromuscular junctions in the tibialis anterior muscles of affected mice, stained for presynaptic synaptophysin and postsynaptic acetylcholine receptors, were of normal size. Quantitation revealed no evidence of nerve terminal retraction or denervation (Fig. 2 and Supplementary Figure S1).
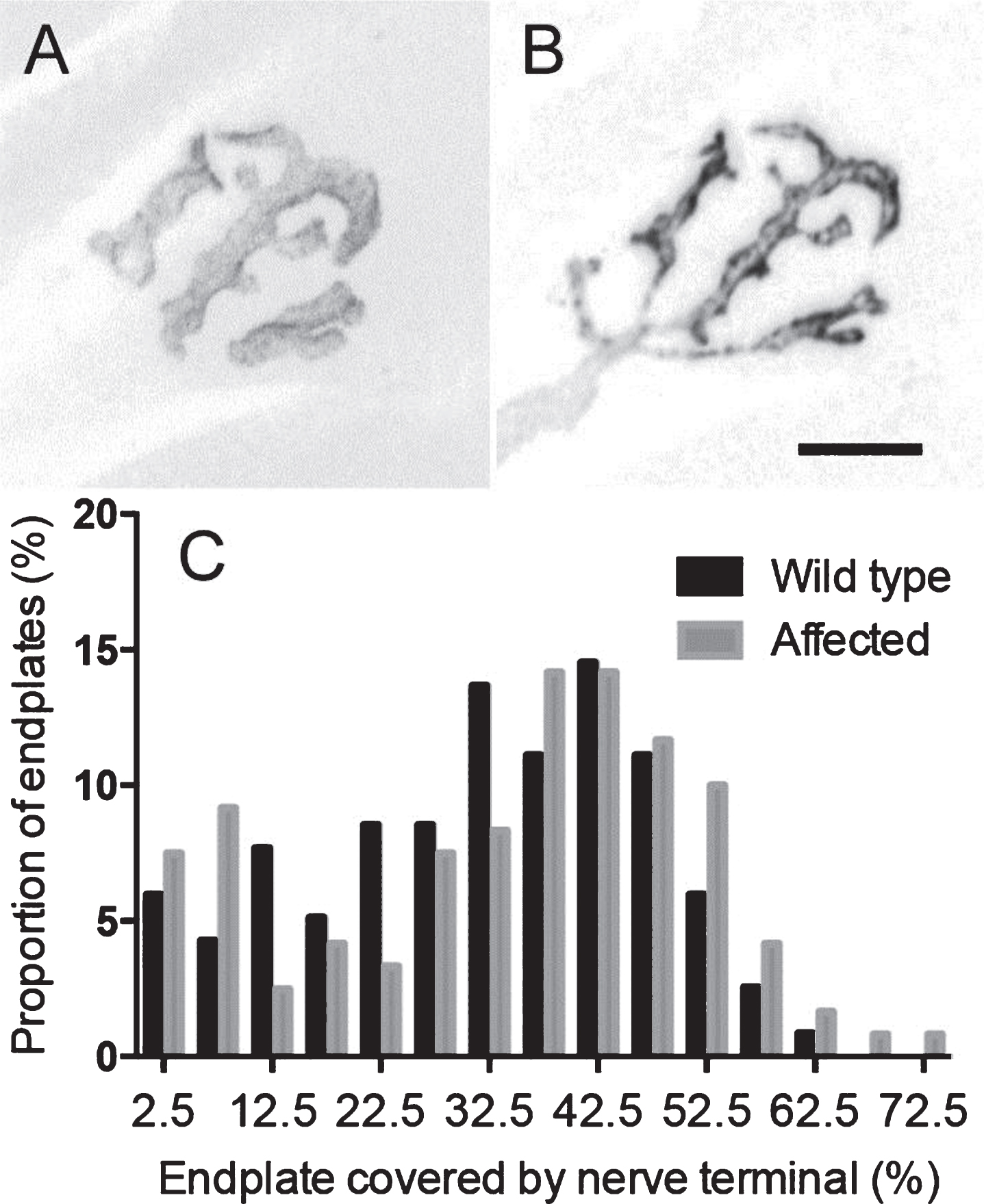
Motor endplates of affected L25 mice remain innervated at the ethical termination. Confocal Z-projection images of an exemplar neuromuscular junction from the tibialis anterior muscle of an L25 mouse. (A) Postsynaptic acetylcholine receptors. (B) Synaptophysin immunostaining of synaptic vesicles in the overlying nerve terminal. Images have been inverted (dark on a white background), and brightness adjusted to improve reproduction. Scale bar = 10 μm. (C) Frequency histogram showing the portion of acetylcholine receptor area at each endplate that was covered by nerve terminal synaptophysin labeling at the time of ethical termination (n = 117, 120 endplates respectively pooled from 6×L25 mice and 6×wild-type mice).
Following whole genome sequencing and subsequent alignment to the mm10 mouse reference genome, the transgene was found to be inserted between chr7:27371152-27390253, resulting in disruption of an intronic region of the Sptbn4 gene, which encodes βIV spectrin. Western blotting of wild-type brain lysates revealed a band of approximately 160kD, representing the Σ6 isoform of βIV spectrin [2]. Additional bands presumably represent other splicing forms of βIV spectrin. By contrast affected L25 transgenic progeny (presumptive L25+/+) revealed no βIV spectrin bands. Unaffected L25 transgenic progeny (presumptive L25+/–) showed bands of diminished density, compared to wild-types (Fig. 3). Blots probed with non-immune IgG2b, as a control for non-specific binding of the primary antibody, revealed no bands (Supplementary Figure S2).
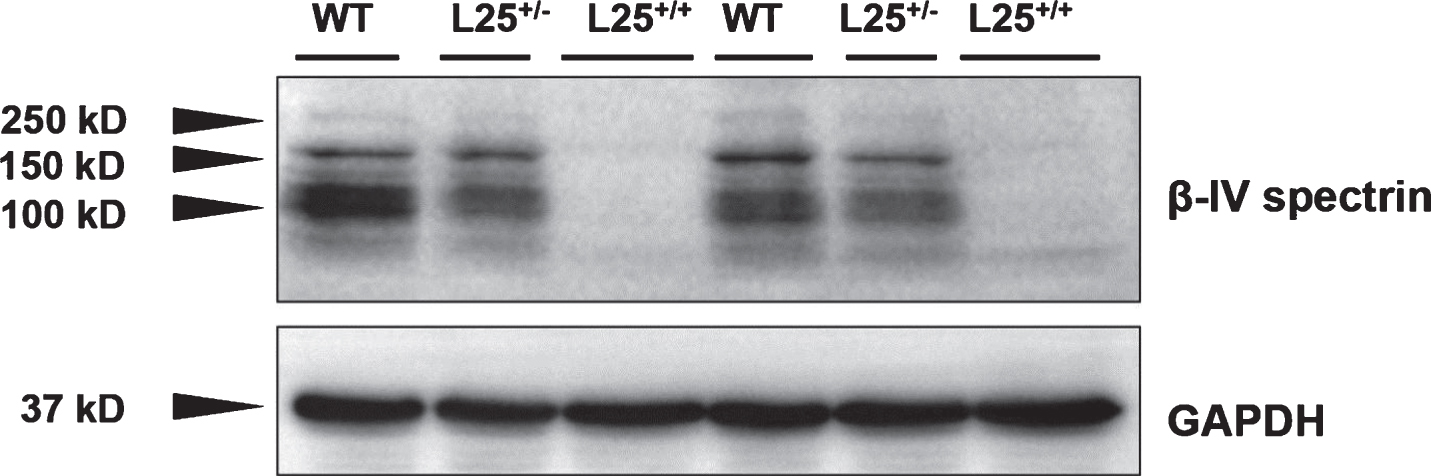
Representative Western blot of brain lysates from wild-type (WT), unaffected transgenic (L25+/–) or affected transgenic (L25+/+) mice probed for βIV spectrin. Wild-type brain lysates reveal strong labelling for βIV spectrin, while brains of the affected (L25+/+) mice showed no bands. Diminished bands are evident for L25+/– mouse brains. GAPDH labeling of the same samples demonstrated equal loading and transfer of protein. Results are shown for two mice of each genotype.
DISCUSSION
In this study we tracked the post-weaning development of a motor phenotype that arose in mice after random insertion of a transgene into the genome. The transgene was later found to have disrupted the β-IV spectrin gene, which was confirmed by reduced expression of βIV spectrin protein in brain homogenates. The motor phenotype was recessive, occurring in 20% of L25+/–×L25+/– progeny. In an earlier study deliberate, targeted inactivation of the βIV spectrin gene resulted in tremor, hearing failure and spasticity, with impaired organisation of the zones of action potential generation/regeneration at the axon initial segment and Nodes of Ranvier [2, 3]. While we did not notice obvious hearing problems in the affected animals, the motor problems we observed support the important role of βIV spectrin for maintaining control of the hind limbs during postnatal maturation. Weekly tracking of phenotype development in the affected L25 mice (conducted blind to genotype) revealed measures of motor impairment that might be useful in future mouse studies of ataxia and other inherited neuro-motor diseases.
While it is important to consider the alternative possibility that ectopic expression of the Sef-b transgene in the brain or spinal cord might have caused the motor phenotype, this seems unlikely. The lens-specific expression cassette used (chick δ1-crystallin enhancer fused to a modified mouse αA-crystallin promoter) was previously shown to target the lacZ marker gene specifically to the eyes of transgenic mouse embryos, with no expression visible in the brain, spinal cord or other tissues [4]. While we cannot exclude the possibility of some leaky expression of Sef-b in the postnatal CNS, line 25 (L25) was just one of a total of six independent mouse lines generated by random genomic insertion of the lens expression cassette, driving either Sef (lines L22-L24) or Sef-b (lines L25-L27). Sef itself is a potent inducer of FGF-signalling, but the Sef-b isoform lacks the signal peptide and is localized within the cytoplasm. The Sef transgenic lines developed persistent and reproducible microphthalmia through adulthood, linked to suppression of FGF-signaling specifically in the lens, but no other phenotype [1]. In contrast, the three Sef-b lines showed no ocular phenotype, consistent with no effect on FGF-signaling in lens. Among the six transgenic lines generated, only L25 displayed a motor phenotype, consistent with the idea that chance disruption of the β-IV spectrin gene (not ectopic Sef-b-activity), was specifically responsible for the motor phenotype.
Disruption of the βIV spectrin gene seems to preferentially impair central motor pathways rather than peripheral motor axons [2, 3]. Consistent with this, the affected L25 mice displayed mainly spasticity and tremor. The paralysis we observed was spastic, rather than flaccid. Motor endplates remained fully innervated (Fig. 2). At the endpoint all the affected mice still displayed the hind limb retraction sign when lifted by the tail. At the endpoint 7/9 males displayed fine tremor, whereas the females did not. One male also displayed a jerky uncontrolled movement. Seven of the mice, including all 5 females, displayed severe hind limb spasticity with balling of the feet that impaired voluntary mobility. Thus the transgene insertion produced varying degrees of tremor and spasticity. Together these results suggest that the predominant effect of the L25 insertion mutation was to impair central motor pathways controlling hind limb movements, while spinal motor neurons retained their peripheral connections to muscle.
Robust quantitative measures of disease progression and endpoint are important for preclinical studies in rodent models of neuromuscular diseases. In models of amyotrophic lateral sclerosis, progression is assessed with measures such as weight loss, grip strength, wire hang time, gait analysis and neurological scoring. A delayed righting response generally serves as the endpoint criterion [6]. Affected L25 mice displayed abnormally low spontaneous motor activity from weaning onward (compared to wild-type littermates). Impaired muscle use and reduced feeding might help explain their poor weight gain in the subsequent postnatal weeks (Fig. 1I-L). Of the three pre-specified endpoint criteria in our study, loss of body weight (≥15%) was what decided termination endpoint in every affected L25 mouse. An accelerating fall in body weight generally occurred in the one or two weeks immediately preceding termination. Body weight also provided a robust endpoint determinant in mouse models of anti-MuSK myasthenia gravis, which instead involves flaccid paralysis [5]. Some studies in the transgenic SODG93R mouse model of amyotrophic lateral sclerosis have used 30% weight loss (over 72-hours) as one of several termination criteria (12). Such severe weight loss would not be ethically acceptable in our jurisdiction. As a measure of impairment, body weight has several advantages; it is non-invasive, easy to measure and objective. As a continuous measure it is suitable for parametric statistics. Body weight loss ≥15% might provide a sensitive endpoint for preclinical mouse models in diverse neuro-motordisorders.
The current study highlights the importance of the β-IV spectrin gene for inhibitory control of lumbosacral motor neurons and suggests the need to screen DNA sequence data of hereditary spastic paraplegia patients for possible mutations in the orthologue.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
Footnotes
ACKNOWLEDGMENTS
The authors thank Drs Michele Gerke, Haydn Allbutt, Louise Cole (Bosch Institute Advanced Imaging Facility) and Jessica Boros and members of the Leamey lab for technical support and advice. This work was supported by MND Australia (Leadership grant) and NHMRC (1095215) to IPB and by University of Sydney Bridging grants to WDP.