
Abstract
Huntington’s disease (HD) is a devastating neurodegenerative disorder characterized by impaired motor function and cognitive decline, ultimately leading to death. HD is caused by a polyglutamine expansion in the N-terminal region of the huntingtin (HTT) protein, which is linked to decreased HTT turnover, increased HTT proteolysis, increased HTT aggregation, and subsequent neuronal death. In this review, we explore the mechanism of the protective effect of blocking HTT proteolysis at D586, which has been shown to rescue the HD phenotype in HD mouse models. Until recently, the mechanism remained unclear. Herein, we discuss how blocking HTT proteolysis at D586 promotes HTT turnover by correcting autophagy, and making HTT a better autophagy substrate, through post-translational myristoylation of HTT at G553.
INTRODUCTION
Huntington’s disease (HD) is a neurological disorder characterized by the irreversible, progressive degeneration of neurons which leads to a combination of motor, psychiatric, and cognitive symptoms. In patient brains, there is extensive degeneration of the striatum, coupled with a comparatively lesser atrophy of the cerebral cortex. Involuntary motor movements and impaired voluntary movements are some of the most salient traits of the disease. However, psychiatric and cognitive symptoms may manifest years before the onset of HD in a prodromal phase. HD is typically adult-onset and spans a period of 15–20 years, ultimately culminating in death. Despite ample research, there are currently no disease-modifying treatments available [1].
HD is caused by a mutation in a single gene that is inherited in an autosomal dominant pattern [2]. The huntingtin gene (HTT), located on chromosome 4, consists of 67 exons and encodes the huntingtin protein (HTT), which spans 3144 amino acids (aas) [2–4]. HD is caused by a CAG repeat expansion in exon 1 of HTT that encodes a polyglutamine (polyQ) expansion near the N-terminus of HTT (Fig. 1) [1, 2]. The expansion is partially penetrant with 36–39 CAG repeats and fully penetrant with 40 or more repeats [5]. In addition, CAG repeat length is inversely correlated with age of disease onset, with longer repeat lengths associated with an earlier age of onset [6–9]. The polyQ expansion induces a toxic gain-of-function, partly attributed to the aggregation of mHTT, which is a hallmark of HD [10–12]. Furthermore, the expansion exerts a toxic loss-of-function of HTT [10, 11], which is involved in many processes [13], including intracellular trafficking [14], cell spindle assembly [15], transcription [16], and endocytosis [17]. More recently, HTT has emerged as a vital player in regulating autophagy [18]. The toxicity of the polyQ expansion is well-established, due to its relevance to HD pathology. However, numerous domains and post-translational modifications (PTMs) downstream of the N-terminal region have also been characterized in wild-type HTT (wtHTT) and mutant HTT (mHTT) and play various roles in function and toxicity. For instance, HTT houses several HEAT (huntingtin, elongation factor 3, protein phosphatase 2A, TOR1) domains that play a crucial role in protein– protein interactions, which are critical to the scaffolding function of HTT in autophagy [19–21].
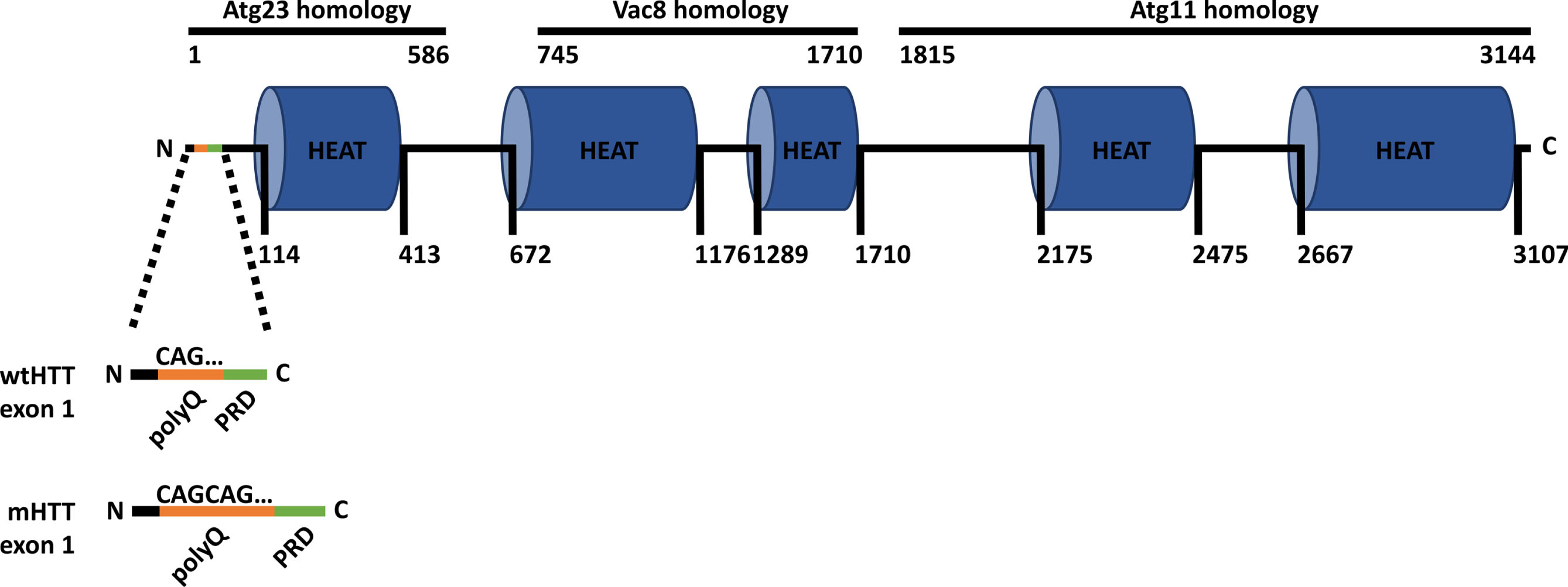
Huntingtin structure. Schematic of the huntingtin protein (HTT) structure, highlighting exon 1 in both wild-type HTT (wtHTT) and mutant HTT (mHTT). The polyglutamine (polyQ) stretch in orange, comprised of CAG triplet repeats, distinguishes mHTT with its expanded polyQ region from wtHTT. The proline-rich domain (PRD) is in green. HTT shares similarities with yeast proteins Atg23, Vac8, and Atg11. The N and C labels indicate the N- and C-terminus, respectively. Numbers indicate the amino acid length.
CATCH 22: AUTOPHAGY IS REQUIRED FOR mHTT CLEARANCE, BUT RELIES ON HTT FUNCTION
Autophagy is the major mechanism of degradation in the cell and is required for the removal of damaged organelles and protein aggregates like mHTT (Fig. 2A) [22]. Autophagy relies on autophagy-related proteins and receptors, including microtubule-associated protein 1A/1B-light chain 3 (LC3) and sequestosome-1 (p62/SQSTM1), which facilitate the clearance of selectively ubiquitinated protein aggregates [22–25]. Autophagy begins with LC3 localization to the phagophore double-membrane, where it recruits p62 bound to ubiquitinated cargo destined for degradation. The phagophore expands and encloses the cargo, fusing to form an autophagosome, which subsequently merges with a lysosome to form an autophagolysosome, where degradation with hydrolases takes place [22]. More recently, wtHTT has been established as a scaffold in autophagy, playing multiple crucial roles in basal autophagy [25–31]. Notably, wtHTT interacts with p62, promoting p62 binding to ubiquitinated cargo and to LC3, which supports that wtHTT participates in the cargo-loading step of autophagy [27]. In addition, wtHTT interacts with ULK1 and Beclin 1, thereby freeing ULK1 from negative regulation by mTOR and facilitating the deubiquitination of Beclin 1. Consequently, these interactions enable ULK1 and Beclin 1 to serve as key initiators of autophagy, underscoring the involvement of wtHTT in the activation of autophagy [27, 30]. In turn, autophagy is required for the removal of mHTT aggregates. However, autophagy is dysfunctional in HD in which cargo-loading defects and decreased autophagosome-lysosome fusion results in an accumulation of empty autophagosomes, thereby leading to a build-up of toxic cellular debris in the cytoplasm of neurons (Fig. 2B) [32–34]. The defective autophagy found in HD is likely a result of the toxic loss-of-function of HTT as a scaffold in autophagy, particularly in the cargo-loading and the activation steps of autophagy.
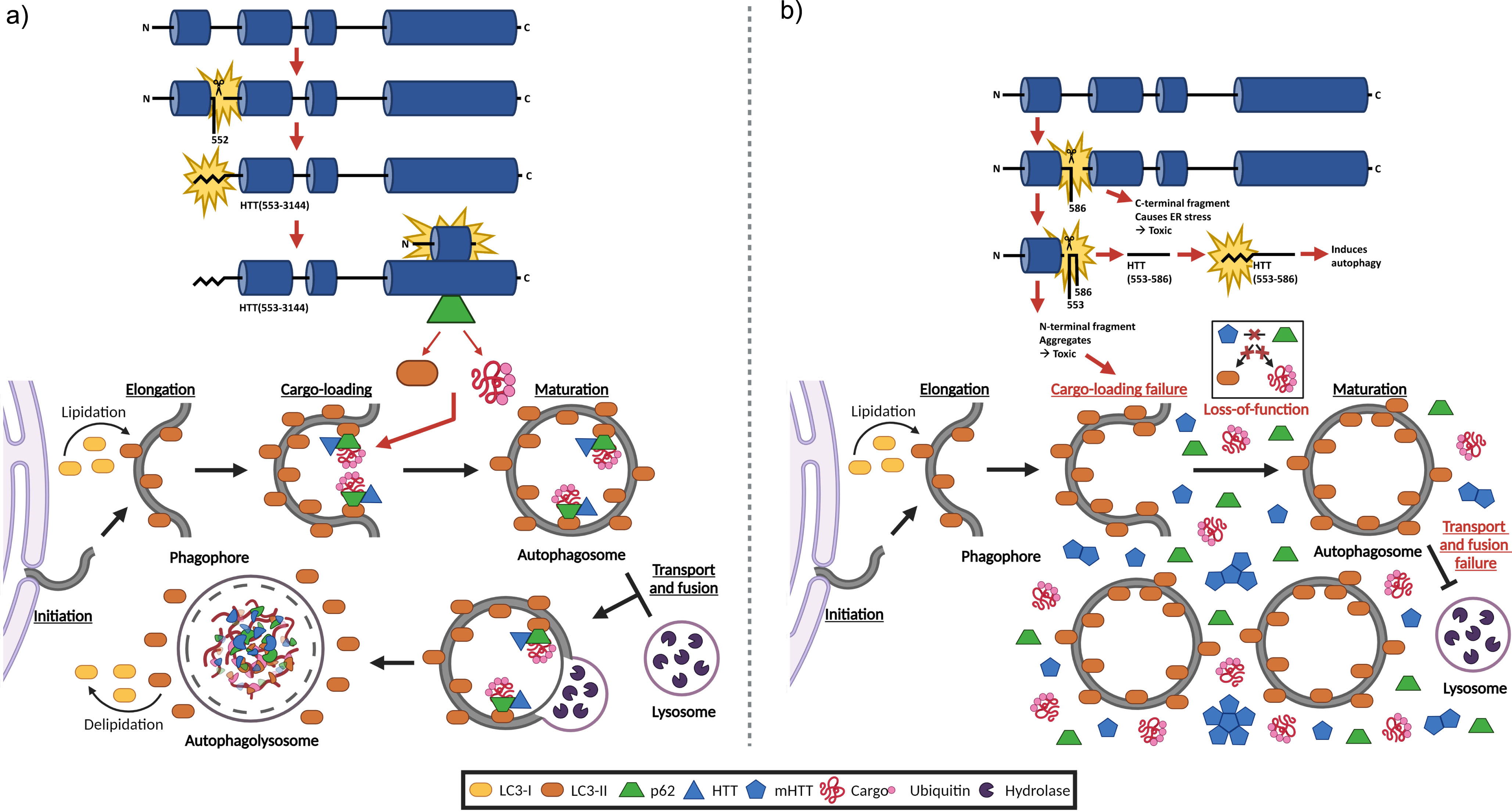
Healthy autophagy versus autophagic dysfunction in HD: Blocking proteolysis of mHTT at D586 restores autophagy. a) Schematic of the model for the protective pathway mediated by inhibition of proteolysis at D586. When mHTT proteolysis at D586 is blocked, proteolysis is redirected to D552, yielding a C-terminal fragment that is subsequently myristoylated at G553. This myristoylated C-terminal fragment interacts with the N-terminal fragment, forming a “full-length” HTT complex that restores autophagy by promoting autophagosome formation and ameliorating the delivery of p62-bound HTT complex to autophagosomes. Consequently, there is increased degradation of interacting HTT fragments through autophagy, resulting in fewer isolated HTT fragments that induce neuronal toxicity. b) Schematic of the model for the disruptive pathway mediated by proteolysis at D586. In HD, full-length mHTT is cleaved at D586, yielding a C-terminal fragment that induces endoplasmic reticulum stress, leading to neuronal toxicity. Further proteolysis at D552 results in an N-terminal fragment that is prone to aggregation, disrupting the autophagic process and contributing to neuronal toxicity. The HTT553 - 586 fragment is then myristoylated at G553, producing a myristoylated fragment that induces autophagy. In HD, autophagy is perturbed at various stages, including dysfunctional cargo-loading, autophagosome transport, and autophagosome-lysosome fusion, resulting in an accumulation of proteins, particularly of mHTT, and empty autophagosomes in the cytoplasm. Figure created with Biorender.
The first links to HTT, HD, and autophagy were shown by the DiFiglia group, whereby HTT aggregation induces the activation of autophagy [35, 36]. Subsequently, Dr. Joan S. Steffan put forward the intriguing hypothesis that HTT evolved from three yeast autophagy-related proteins: Atg23, Vac8, and Atg11 [37]. Later, in the Berthiaume group, we identified a new autophagy-inducing domain within HTT, confirming HTT’s involvement in autophagy [29]. Steffan’s theory was subsequently established by her own group in an elegant study using fly and yeast models detailing that the N-terminal region of HTT (aas 1-586) is homologous to the Atg23 yeast protein, the central HTT region (aas 745-1710) is homologous to Vac8, and the C-terminal region (aas 1815-3144) is homologous to Atg11 [26]. Furthermore, they demonstrated that HTT performs cellular functions analogous to those carried out by these three yeast proteins [26]. In yeast, Vac8 is part of the Atg1 complex that plays a key role in initiating autophagy, while Atg23 and Atg11 work together to regulate autophagosome formation. In mammals, the N-terminal region of HTT, akin to Atg23, interacts with the C-terminal region, akin to Atg11 [26, 37]. Interestingly, it has been shown that interactions between the N- and C-terminal regions are disrupted upon mHTT proteolysis at multiple sites, and promoting the interaction between these two regions can have protective effects [38].
In addition to proteolysis, HTT undergoes numerous diverse PTMs that regulate different aspects of HTT function [39, 40], including ubiquitination [41], phosphorylation [42], SUMOylation [43], methylation [44], S-acylation [45], acetylation [46], and proteolysis [47]. These PTMs can be greatly impacted by the presence of the HD mutation, thereby contributing to the HD phenotype. Furthermore, there exists a dynamic interplay among various PTMs of mHTT, wherein these modifications influence one another, potentially offering protective effects or further contributing to pathogenesis [39]. For instance, phosphorylation at S13 and S16 is protective in HD mouse models, improving behavioral dysfunction, reducing mHTT aggregates, as well as preventing selective neurodegeneration of the striatum and cortex [48]. Similarly, S421 phosphorylation ameliorates behavioral impairment and striatal neurodegeneration in HD mouse models [49], potentially by restoring the axonal transport of brain-derived neurotrophic factor (BDNF)-containing vesicles [50]. Furthermore, phosphorylation of mHTT at S421 is decreased in HD mouse models, and restoring this phosphorylation has also been shown to reduce mHTT toxicity by inhibiting proteolysis at D586 in cells [51, 52]. Therefore, mapping the HTT PTM network is essential for identifying rate limiting PTMs that play protective roles by regulating multiple downstream PTMs, HTT function, and significantly impacting the pathogenesis and phenotypeof HD.
FRIEND OR FOE? PATHOGENIC PROTEOLYSIS OF HTT
A critical event in the pathogenesis of HD involves the proteolysis of full-length mHTT at multiple sites (Table 1), resulting in the generation of N- and C-terminal fragments that exhibit toxicity towards neurons via independent pathways [38, 54]. HTT undergoes proteolysis through the action of proteases, predominantly caspases, and to a lesser extent, calpains [47, 56]. HD patient brains show elevated protease activity, accompanied by the ensuing accumulation of N- and C-terminal fragments [13, 57]. Concurrently, wtHTT can be cleaved at multiple sites in healthy brains and some of the fragments produced are highly toxic [38, 58].
Primary HTT proteolysis sites
Active sites are primary sites of proteolysis, whereas silent sites are not believed to be cleaved.
mHTT proteolysis at D586 is crucial for disease progression in HD mouse models [59], and is likely one of the rate limiting PTMs where upstream PTM effects converge to regulate HD pathogenesis. Proteolysis at D586 is followed by proteolysis at D552, exposing the N-terminal glycine at G553, which is post-translationally myristoylated (Fig. 2B) [25, 29]. This process generates a myristoylated HTT553 - 586 fragment that promotes autophagosome formation through a mechanism that is not fully understood. One theory suggests that the myristoylated HTT553 - 586 fragment detects and induces membrane curvature at the endoplasmic reticulum (ER), which is the origin site of phagophores. This preposition stems from the observation that the myristoylated HTT553 - 586 fragment exhibits sequence homology with the Barkor/ATG14 L autophagosome-targeting sequence (BATS) domain of ATG14 L, which senses and promotes membrane curvature at the ER, thereby initiating the formation of autophagosomes [25, 60].
Myristoylation involves an irreversible, covalent attachment of a saturated 14-carbon myristic acid to the N-terminal glycine residue of proteins through an amide bond (Fig. 3) [61]. This process is catalyzed by two N-myristoyltransferases (NMTs) and takes place either co-translationally, following the removal of the initiator methionine residue, or post-translationally, following the exposure of a previously internal glycine residue by means of proteolytic cleavage [61, 62]. Myristoylation of HTT at G553 occurs post-translationally, as the N-terminal glycine is exposed by means of proteolysis at D552.
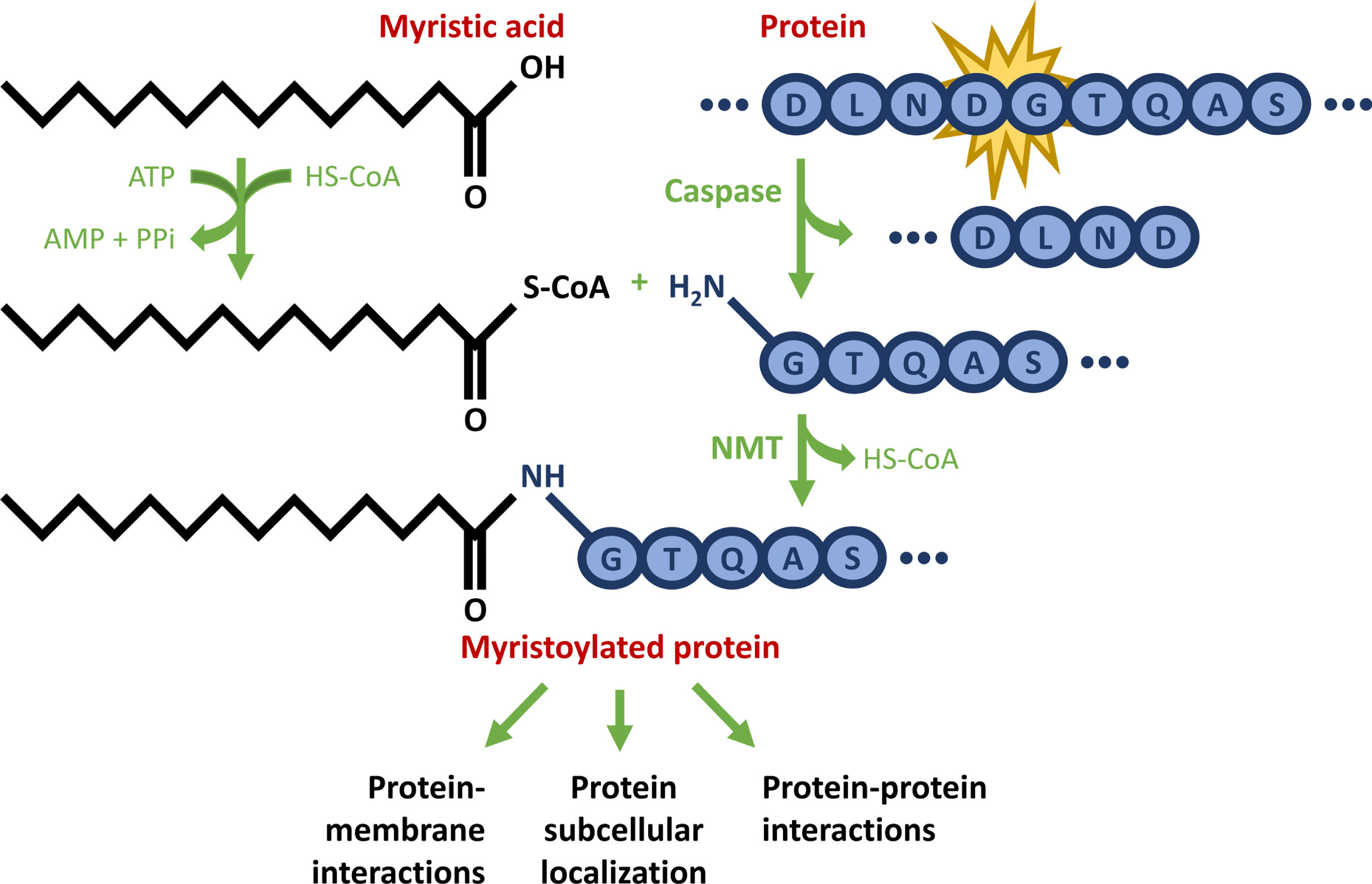
Schematic representation of post-translational myristoylation of HTT. First, the protein undergoes caspase cleavage to reveal a glycine residue. Concurrently, a saturated 14-carbon myristic acid is converted to myristoyl-CoA. Ultimately, myristoyl-CoA is covalently added to the newly exposed N-terminal glycine residue via an amide bond, catalyzed by N-myristoyltransferase (NMT), thereby providing a novel function for proteolyzed proteins. This example shows the HTT amino acid sequence D549-S557.
FRIEND OR FOE? PROTECTIVE PROTEOLYSIS OF HTT
When expressed separately, both the N-terminal HTT1 - 552 and C-terminal HTT587 - 3144 fragments display neuronal toxicity (Fig. 2B) [38]. The N-terminal HTT1 - 552 fragment forms aggregates and induces neuronal toxicity through several mechanisms, including impairment of autophagy [36, 54]. In contrast, the C-terminal HTT587 - 3144 fragment induces neuronal toxicity by inactivating dynamin 1 at ER membranes, leading to ER stress, ultimately leading to cell death [38]. Interestingly, proteolysis at a single site does not lead to neuronal toxicity, as it still allows the N- and C-terminal fragments to interact, preventing their toxic individual actions, whereas proteolysis of mHTT at multiple sites decreases the interaction between the N- and C-terminal fragments and leads to neuronal toxicity [38]. This suggests that not all proteolysis is toxic, and that the protective mechanism of inhibiting caspase cleavage at D586 works by maintaining the interaction between the N- and C-terminal fragments.
Post-translational myristoylation is a unique modification that facilitates a new function for proteolyzed proteins by promoting novel protein– protein interactions, protein-membrane interactions, and protein subcellular re-localization. This process could shed light on the mechanism by which proteolysis at a single, specific site enables the N- and C-terminal fragments to interact, and allows HTT to function as a scaffold in autophagy— a membrane-driven process [61, 64]. Notably, post-translational myristoylation of HTT is significantly reduced in the presence of the polyQ expansion [25, 65], suggesting compromised protein– protein and protein-membrane interactions, which potentially contribute to the impaired autophagy observed in HD.
Blocking HTT proteolysis at D586 significantly increases proteolysis at D552 and post-translational myristoylation at G553. Essentially, when proteolysis at D586 is blocked, mHTT proteolysis switches to directly target D552, exposing the N-terminal glycine at G553, which is post-translationally myristoylated (Fig. 2A) [65, 66]. This process generates a myristoylated C-terminal HTT553 - 3144 fragment that interacts with the N-terminal HTT1 - 552 fragment; meaning that there are no free N-terminal or C-terminal fragments to induce neuronal toxicity. Of note, myristoylation is vital to the interaction between N-terminal HTT1 - 552 and C-terminal HTT553 - 3144 fragments, as blocking myristoylation through a G553A mutation decreases the interaction between the given fragments [65].
The above scenario could explain the rescue of impaired autophagy in caspase-6-resistant (C6 R) compared to YAC128 mice [31]. C6 R mice, a transgenic HD mouse model with 128 CAG repeats and resistance to caspase-6 proteolysis at D586, do not exhibit accumulation of toxic mHTT aggregates, neurodegeneration, and behavioral phenotypes that are typically observed in the YAC128 HD mouse model [31, 67].
More evidence that post-translational myristoylation of HTT is protective came with the identification of a naturally occurring single nucleotide polymorphism (SNP) that creates a G553E substitution, which abolishes post-translational myristoylation of HTT and increases toxicity. Moreover, the G553E polymorphism blocks proteolysis at D552, redirecting caspase cleavage to the toxic D513 site, after proteolysis at D586 [68]. Proteolysis at D513 results in toxic HTT fragments with decreased interaction, and is thought to contribute to overall cellular toxicity [38, 68]. Interestingly, the G553E substitution occurs on the control allele, rather than the mutant HD allele. Having both the polymorphism and the mutation suggests that proteolysis at D513 and 586 occurs on the control allele, and simultaneously, proteolysis at D586 occurs on the allele with the HTT mutation, resulting in an overall loss of protective myristoylation and an abundance of toxic HTT fragments. This also means that an individual carrying both the G553E SNP and the HD mutation may have an earlier onset of disease [68]. However, no clinical information was available on these patients to assess the potential modifying effects of the SNP. In such cases, blocking proteolysis at D586 in mHTT may not be sufficient to mitigate the HD phenotype in HD patients with the G553E substitution. More research is required to uncover the combined impact of the G553E polymorphism and the HTT mutation on phenotype.
CONCLUSION AND FUTURE DIRECTIONS
Overall, blocking proteolysis at D586 is protective and ameliorates the HD phenotype in HD mouse models. This is linked to the redirection of proteolysis to D552, resulting in myristoylation at G553. Consequently, this facilitates the interaction between the N-terminal HTT1 - 552 and the myristoylated C-terminal HTT553 - 3144 fragments, leading to the formation of a “full length” HTT complex that is a better substrate for autophagy. This “full length” HTT complex rescues autophagy, likely by promoting autophagosome formation while also enhancing the delivery of bound p62 to autophagosomes for their ultimate degradation. This leads to increased breakdown of interacting HTT fragments through autophagy, resulting in fewer isolated N- and C-terminal fragments that induce neuronal toxicity.
The recent development of antisense oligonucleotide (AON) 12.1 to remove the proteolytic site at D586 represents a significant advancement for targeting the D586 proteolytic site [69, 70]. AON12.1 induces the partial skipping of HTT exon 12, removing the sequence encoding the caspase-6 proteolysis site. The resulting protein, referred to as HTTΔ12, lacks the caspase-6 proteolysis site and is resistant to proteolysis at D586. Consequently, HTTΔ12 retains the structural and functional properties of wtHTT and shows an improved HD phenotype in YAC128 mice [70]. This improvement is likely attributed to the diversion of HTT proteolysis to D552, leading to myristoylation of the C-terminal fragment at G553. This results in increased interaction of N- and C-terminal fragments, conferring upon HTTΔ12 the same structural and functional properties as wtHTT, thus restoring autophagy and mitigating the HD phenotype.
Previous attempts focused on inhibiting the primary enzyme responsible for proteolysis at D586, caspase-6 [71, 72]. However, this approach proved difficult likely due to the involvement of caspase-6 in cellular processes essential for proper brain function and apoptosis [70]. In addition, HTT can undergo proteolysis at D586, albeit to a lesser extent, by caspase-8 and -10 [72]. Thus, targeting caspase-6 is not ideal. Interestingly, caspase-1 cleaves and activates caspase-6 [73]. Therefore, inhibition of caspase-1 indirectly inhibits proteolysis at D586 by inhibiting caspase-6 activation [66]. This positions caspase-1 as a potentially more promising target than caspase-6. Caspase-1 plays an important role in initiating the inflammatory response against various stressors. Caspase-1 inhibition has been studied in the context of treating cytokine release syndrome, a life-threatening condition characterized by excessive levels of circulating pro-inflammatory cytokines [74], and Alzheimer disease (AD), a neurodegenerative disorder associated with microglial inflammation [75, 76]. The LeBlanc group linked caspase-1 activation of caspase-6 with axonal degeneration and neuronal inflammation in AD and suggested that a caspase-1 inhibitor is a potential therapeutic for AD [75]. Furthermore, inhibition of caspase-1 using VX-765 (Belnacasan) improved neuropathology in AD mouse models [76]. In HD, neuronal cell death triggers inflammation, which further contributes to neuronal cell death, leading to a positive feedback loop of neurodegeneration and central inflammation. More recently, there is also evidence of peripheral inflammation both in HD patients and mouse models, with caspase-1 likely involved in HD pathophysiology [77, 78]. Consequently, the inhibition of caspase-1 holds the potential to address two critical aspects simultaneously— indirectly inhibiting proteolysis at D586 through inhibition of caspase-1-mediated activation of caspase-6, as well as mitigating inflammation in HD. In addition, the FDA-approved antibiotic and caspase-1 inhibitor minocycline was found to slow disease progression and prolong survival of HD mouse models [79]. Furthermore, minocycline was shown to be tolerable and neuroprotective in HD patients [80–83]. However, minocycline did not provide sufficient evidence to justify a larger and longer trial in HD [84]. As an antibiotic, minocycline is unlikely to provide long term therapeutic effects in HD. Nevertheless, minocycline highlights the potential of more specific caspase-1 inhibitors. Potential future directions may involve exploring the combination of AON12.1 with caspase-1-inhibiting drugs. Utilizing both approaches allows for the directed intervention of HTT at both the mRNA and protein levels, while also addressing the distinctive inflammation associated with HD. This dual-targeting strategy has the potential to significantly enhance the amelioration of the HD phenotype, presenting a promising avenue for HD therapeutics. It offers a comprehensive approach that targets multiple aspects of HD pathology.
Footnotes
ACKNOWLEDGMENTS
We would like to acknowledge that the University of Waterloo resides on the traditional territory of five Indigenous communities, including the Ho-de-no-sau-nee-ga (Haudenosaunee), Mississaugas of the Credit First Nation, Anishinabewaki ᐊᓂᔑᓈᐯᐗᑭ, Attiwonderonk (Neutral), and Mississauga peoples, which is situated on the Haldimand Tract, a land granted to the Six Nations, encompassing six miles on either side of the Grand River. This land is home to many past, present, and future Indigenous peoples.
FUNDING
This work was supported by a Natural Sciences and Engineering Research Council (NSERC) Discovery Grant (RGPIN-2019-04617) to DDOM, Canadian Institutes of Health Research (CIHR) Canada Graduate Scholarship Master’s to YA, University of Waterloo President’s Graduate Scholarship to YA, and University of Waterloo Graduate Scholarship to YA.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.