
Abstract
Neuroimaging is increasingly being included in clinical trials of Huntington’s disease (HD) for a wide range of purposes from participant selection and safety monitoring, through to demonstration of disease modification. Selection of the appropriate modality and associated analysis tools requires careful consideration. On behalf of the EHDN Imaging Working Group, we present current opinion on the utility and future prospects for inclusion of neuroimaging in HD trials. Covering the key imaging modalities of structural-, functional- and diffusion- MRI, perfusion imaging, positron emission tomography, magnetic resonance spectroscopy, and magnetoencephalography, we address how neuroimaging can be used in HD trials to: 1) Aid patient selection, enrichment, stratification, and safety monitoring; 2) Demonstrate biodistribution, target engagement, and pharmacodynamics; 3) Provide evidence for disease modification; and 4) Understand brain re-organization following therapy. We also present the challenges of translating research methodology into clinical trial settings, including equipment requirements and cost, standardization of acquisition and analysis, patient burden and invasiveness, and interpretation of results. We conclude, that with appropriate consideration of modality, study design and analysis, imaging has huge potential to facilitate effective clinical trials in HD.
Keywords
INTRODUCTION
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder caused by a CAG repeat expansion in the first exon of the HTT gene, which encodes a mutant huntingtin protein [1]. Typically, HD is an adult-onset degenerative disease with a protracted period of decline, typically lasting 15 to 20 years. The clinical disease is characterized by a triad of symptoms, including neuropsychiatric disturbance, cognitive decline, and progressive motor dysfunction. The early behavioral and psychological manifestations are subsequently followed by the onset of involuntary movements, along with a simultaneous decline in cognitive function, and various hyperkinetic movements including chorea [2]. All these in combination relentlessly progress and ultimately lead to severe disability and morbidity, and finally a bed-bound state and death.
Currently, there are several potential disease-modifying interventions in development (reviewed by Tabrizi et al. [3]). These include therapies which aim to reduce the toxic agent in the brain, mutant huntingtin (mHTT), by targeting Huntingtin DNA and RNA, and clearance of the huntingtin protein. There are also therapies aiming to modify or modulate DNA repair pathways to address somatic instability of the mutant HTT gene, that likely contribute to the disease pathogenesis [4]. Additionally, there are attempts to modify the HD disease course by addressing additional, likely indirect pathophysiological mechanisms. Therefore, at present, there are many HD clinical trials that are progressing at various stages and paces. Given the heavy unmet need in HD, efficient conduct of clinical trials that leads to scientifically robust conclusions is essential. Many aspects of the clinical trial process can be facilitated by the inclusion of neuroimaging, from participant selection and safety monitoring, through to demonstration of disease modification. For this reason, it is timely to review and discuss the state and roles of neuroimaging in clinical trials of HD.
The European Huntington’s Disease Network (EHDN) is a non-profit research network committed to advancing research, facilitating the conduct of clinical trials, and improving clinical care in HD. One of the ways the EHDN achieves this is through a network of working groups, each addressing a specific need (www.euro-hd.net/html/network/groups). The focus of the Imaging Working Group is to openly discuss, develop and promote best practices for the acquisition and analysis of neuroimaging data in HD. One of our key objectives is to facilitate the use of neuroimaging in clinical trials. As a step towards achieving this objective, a sub-team of the EHDN Imaging Working Group was formed and tasked with writing a current opinion article on the use of imaging in HD trials. The sub-team was composed of both industry and academic experts across the key imaging modalities: volumetric-, diffusion- and functional-MRI and perfusion imaging, positron emission tomography (PET), magnetic resonance spectroscopy (MRS), and magnetoencephalography (MEG). We structure our discussions around six key areas, particularly relevant for clinical trial planning and execution (adapted from Schwarz et al. [5]). The areas are: Neuroimaging to aid participant selection, enrichment, stratification, and safety monitoring Neuroimaging as a tool to demonstrate biodistribution, target engagement, and pharmacodynamics Neuroimaging as a tool to provide evidence for disease modification Neuroimaging as a tool to understand brain re-organization following therapy Challenges for translation to clinical trials Multi-modal neuroimaging to aid interpretation of trial data
We hope that this article will provide a useful resource tool for those planning to include neuroimaging in future HD trials. First, we briefly describe the imaging modalities that are included in the review. The main usage of the different types of imaging is summarized in Table 1.
Summary of image types and their main usage in HD research
Structural/volumetric MRI
Structural magnetic resonance imaging (sMRI) is a non-invasive technique for visualizing brain anatomy and quantifying its macrostructure, with good spatial resolution. While it is standard to obtain 3D T1-weighted images at 1×1×1 mm isotropic resolution (preferably at 3 Tesla (T)), even finer resolutions are possible but not commonly used in clinical trials.
MRI employs powerful superconducting magnets and pulsating radio frequency (RF) currents to provide information on the tissue being scanned [6]. By varying the sequence of RF pulses applied and the resonance collected, different types of images can be constructed. The most commonly acquired sequences are 3D T1-weighted and 2D/3D T2-weighted MRI. These acquisitions vary in terms of contrast and brightness (see Fig. 1). Briefly, T1 (longitudinal relaxation time) and T2 (transverse relaxation time) control tissue contrast. Short T1 tissues (e.g., fat, where the tissue quickly realigns its longitudinal magnetization with the main magnetic field) are bright on T1 images and short T2 tissues are dark on T2 images; cerebrospinal fluid (CSF) is dark on T1-weighted images and bright on T2-weighted images (Fig. 1; see [7] for a more detailed description). T1-weighted MRI scans are excellent for the delineation and quantification of brain macrostructure because of their well-defined anatomical boundaries within the brain. T2-weighted MRI scans are considered best suited for detection of pathology. A third commonly acquired structural sequence is the fluid attenuated inversion recovery (FLAIR) scan. This has similar properties to the T2-weighted scan but with longer times between successive pulse sequences (Repetition Time, TR) and between the delivery of the RF pulse and the receipt of the echo signal (Echo Time, TE). The CSF signal is suppressed on FLAIR scans, appearing dark, whilst abnormalities are highlighted as bright (hyperintense). This makes FLAIR scans more sensitive to pathology since the differentiation between CSF and abnormalities is clearer. FLAIR scans are an essential part of MRI safety evaluations in clinical trials. T2*-weighted scans, which show hemorrhages and hemosiderin deposits as dark (hypointense), are often additionally used as part of safety evaluations.
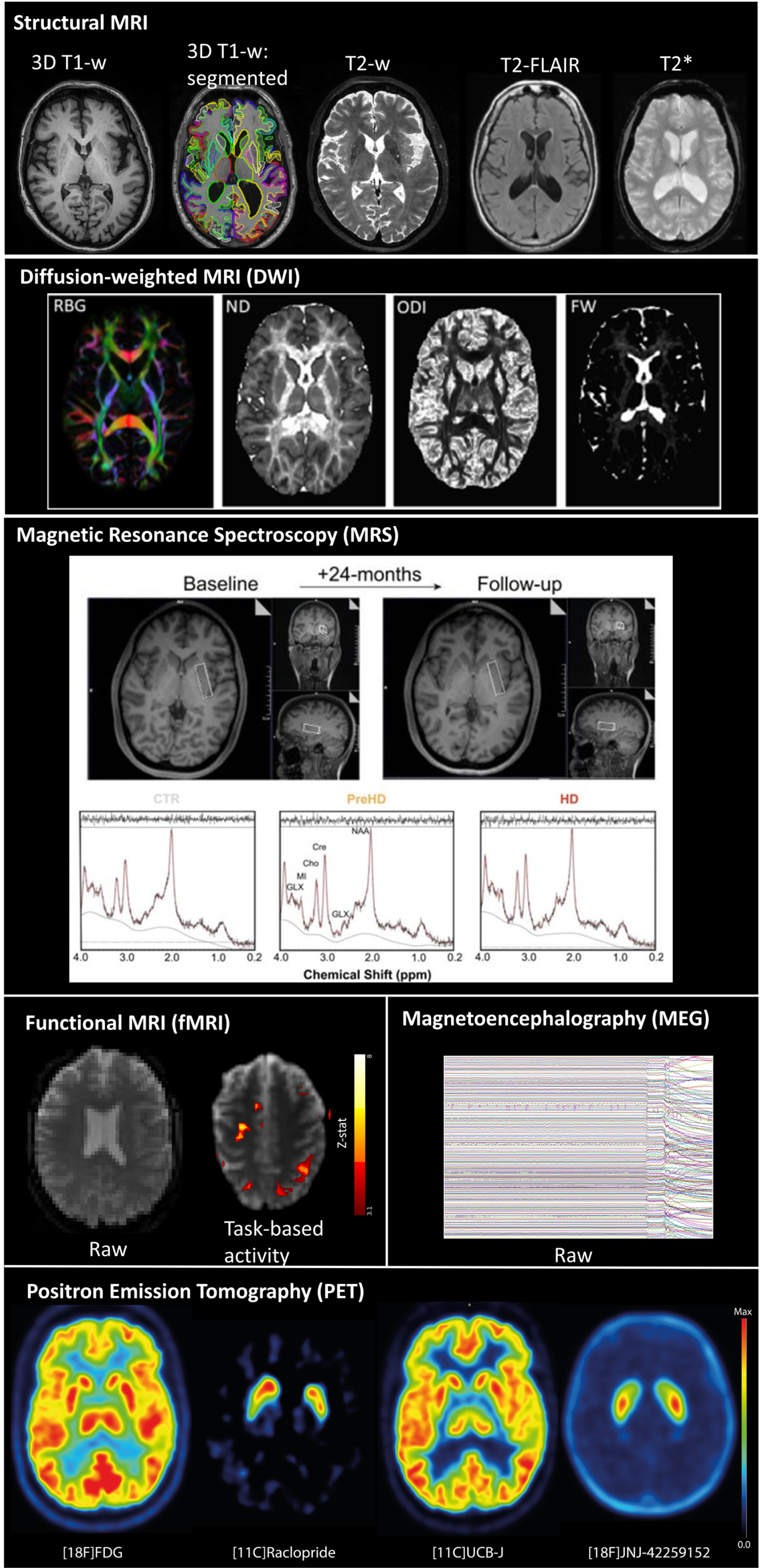
Quantification of T1-weighted scans typically includes regional grey matter, white matter, and CSF volumes. These can be obtained at baseline and as volume change over time, described as atrophy in neurodegenerative diseases if longitudinal (repeat) scanning is performed (Table 1). Regional cortical thickness can also be quantified at baseline and longitudinally; however, cortical thickness measures are less commonly used in trials compared with volume/atrophy measurements, due to a lack of confidence in their reliability over certain cortical regions [8]. In terms of application to clinical trials, sMRI is the most mature imaging modality. Even so, appropriate site set-up, rigorous quality control for hardware, acquisition steps and image reconstruction, as well as careful consideration of the analysis process, are imperative for obtaining reliable data (discussed in Sections 1 and 5).
Diffusion MRI
Diffusion weighted imaging (DWI) is a non-invasive technique, providing information about brain microstructure and has been most widely used to interrogate white-matter integrity (described in detail by Soares et al. [9]). This acquisition highlights the diffusivity of water within the brain. There is generally high diffusivity in fluid-filled spaces such as the CSF; when water molecules are able to move freely in all directions. This is known as isotropy. In contrast, in highly organized structures such as the corpus callosum, diffusion is much more constrained and tends to be directional, i.e., is anisotropic. Usually, DWI acquisitions in HD clinical trials use a single b-value (1000 s/mm2) for diffusion-weighting (“single shell”) and a diffusion tensor imaging (DTI) model to characterize extracellular diffusion patterns as indices of brain tissue microstructure. Analyses of DWI data with DTI typically generate the following metrics: fractional anisotropy (FA) (a measure of diffusion directionality), mean diffusivity (MD), axial diffusivity (AD) (along the principal fiber direction), and radial diffusivity (RD) (perpendicular to the principal fiber direction) (see review by Estevez-Fraga et al. [10]). A loss of structural integrity is generally signified by a decrease in FA and AD and an increase in RD and MD. DTI measures, however, are not only sensitive to biological processes (such as myelin, axon membrane, density, etc.) but also to geometrical features of the fibers (such as fiber crossing/kissing, where FA may drop) making them difficult to interpret in terms of specific biological white-matter properties [11]. More recently, increasingly sophisticated models are being developed to interrogate “multi-shell” DWI data acquired over a range of b-values that allow separate estimation of extracellular diffusivity (at lower b-values) and intracellular diffusivity (at higher b-values). For instance, the neurite orientation dispersion and density imaging (NODDI) [12, 13] and the composite hindered and restricted model of diffusion (CHARMED) [14] provide indices of intracellular diffusion signals as estimates of axon density and have been shown to be sensitive to white-matter neuropathology in HD [15, 16] (Fig. 1). In addition to considering which DWI acquisition to use and which model to apply to generate metrices, there are also different pre-processing techniques to be considered (e.g., eddy current, motion, and geometric distortion correction), as well as a variety of imaging tools for analysis including region- or white-matter tract of interest, whole-brain techniques such as voxelwise tract based spatial statistics (TBSS) [17] or tractography, and graph theory (Table 1). Given that many of the analysis techniques are currently best suited for exploratory analyses in clinical trials, we recommend a priori selection of a method along with precise delineation of statistical thresholding and inferences to be drawn, defining the reasons behind that selection, prior to embarking on statistical data analysis.
Functional MRI and perfusion imaging
Functional MRI (fMRI) is a non-invasive MR neuroimaging method used to indirectly measure brain metabolic and neural activity or connectivity during performance of a task (task-fMRI) or at rest (resting-state fMRI). The most common method is blood-oxygen-level dependent (BOLD) contrast [18]. This method measures susceptibility-related changes in MRI signal, which are due to changes in blood oxygenation induced by neuronal activity. Another method that can be used for fMRI is arterial spin labelling (ASL) perfusion [19]. BOLD fMRI has higher signal-to-noise ratio and better temporal and spatial resolution than ASL. The main use of ASL is the non-invasive quantification of cerebral blood flow (CBF). In ASL, arterial blood water is magnetically labelled using RF pulses while the patient is lying within a strong static magnetic field. As the labelled blood travels to the brain, it gradually loses the longitudinal magnetization induced by the RF pulses and it is this decay that is measured as ASL signal. To quantify CBF, a labelled image is compared against an unlabeled, controlimage.
PET
Positron emission tomography (PET) is a quantitative and highly sensitive imaging technique. Injection of a PET radiotracer aimed at a specific target (i.e., glucose metabolism, receptor, enzyme, protein deposit) enables the visualization and quantification of functional or molecular properties of processes in vivo. 11C and 18F are frequently used radionuclides to label small molecule PET tracers for brain imaging. These radionuclides decay at known decay constants, and the coincident photons emitted after annihilation of the positrons thus emitted, are identified by a ring of detectors in the PET scanner [20]. After acquisition, PET images are corrected for decay, scatter, randoms and deadtime. Because brain volume loss is often observed in neurodegenerative disorders such as HD, and the spatial resolution for state-of-the-art commercial total body PET scanners is 3–4 mm, PET data can be corrected for partial volume effects using structural MR information [21].
Brain PET imaging is highly valued in neuroscience to study neuropsychiatric disorders, including HD, in vivo. Quantitative brain PET tracers can serve as objective biomarkers to study disease pathophysiology, measure disease progression, sample enrichment and monitor novel therapeutics in clinical trials, as outlined in this current opinion article. A range of PET ligands have been used in neurodegenerative conditions including measures of brain activity determined by glucose metabolism (e.g., [18F]FDG PET), or those binding to specific receptors such as D2/3 ([11C]raclopride), cannabinoid (e.g., [18F]MK-9470) and PDE10A (e.g., [11C]IMA107) or proteins such as translocator protein (TSPO), indicating microglial activation as a marker for neuroinflammation. PET radioligands for mHTT are currently in clinical development. The ability to directly target the toxic agent in HD would present a significant advancement in the field.
MRS
Magnetic resonance spectroscopy (MRS) is another versatile MR imaging technique that enables the assessment of metabolic changes in the brain. By combining the ability to differentiate molecules with MRI’s features of localization for the measurement of component chemistry in a given tissue, MRS allows unique insights into brain metabolic or disease (pathophysiologic) processes [22], or the quantification of specific molecules [23]. It has been used to explore the biochemical changes that occur in HD and allows the identification of the relationships of those changes with disease progression or other outcome measures. The concentrations of metabolites such as N-acetylaspartate (NAA) or myo-Inositol (MI) are identified from spectral peaks and the degree of chemical shift is expressed in part-per-million (ppm). Given recent and ongoing advances in technology and methodology, and provided the required expertise is available at the study sites, MRS can be used in HD clinical trials. The window MRS provides into the biochemical composition of brain tissues is difficult to achieve through other imaging modalities, except perhaps with PET.
MEG
Magnetoencephalogram (MEG) is a non-invasive functional neuroimaging technique which measures the extremely small magnetic fields generated by firing neurons. Compared with electroencephalography (EEG), it is less susceptible to noise from the intervening tissues of the skin and skull [24]. MEG can detect magnetic signals generated by a group of ∼50,000 pyramidal neurons from a cortical area of approximately 1 cm2, with extremely high temporal resolution, in the millisecond precision. This is in contrast to functional MRI which captures hemodynamic response to changes in local electrical activity, and thus has latency and longevity of up to 5 seconds [25]. Therefore, MEG has potential to track progressive cortical neuronal loss in HD. Although primarily sensitive to cortical function, MEG can investigate deeper structures using the beamformer technique [26]. This involves extracting the signals from specific regions of interests, including deep subcortical structures such as the striatum. The theoretical spatial resolution using this method is in the sub-millimeter level.
Currently, there is a scarcity of MEG neuroimaging studies in HD [27]. Further work in this domain is essential before MEG can be considered for implementation in HD trials; however, in this review article, we discuss the potential of MEG in HD and lessons learnt from other neurodegenerative disorders.
Statistics in imaging
The essential steps of a neuroimaging study, no matter what the modality may be, include the image processing pipeline and statistical modelling. In the case of image processing, besides converting original data from different types of detectors (collimators in the case of PET to RF receiving coils in MRI), care must be exercised to correct any artifacts that may have been introduced to the data due to head motion, breathing, heart rate, scanner hardware and operator inconsistency, where possible. In time-series analyses, it is recommended to add various additional co-variates in the statistical model. A major goal of statistical modelling is to reduce the massive amounts of raw data to meaningful and concise information that allows the original experimental question to be answered. A very common framework for analyzing 3D images is the mass univariate model, where a regression model is fit separately at each voxel. Inferences over the image are then corrected for multiple testing using random field theory or nonparametric resampling methods (e.g., permutation or bootstrap). Alternative analyses methods like the Boundary Shift Integral [28 –30] may also be used that are region-specific and hypothesis-driven. Given the perpetually growing collection of imaging modalities, and a multitude of pre-processing options, as well as various models that allow for the search of statistical significance, it is essential that detailed analysis plans that address multiplicity, are generated ahead of the experiments, and ideally included as part of study plan pre-registration.
SECTION 1: NEUROIMAGING TO AID PARTICIPANT SELECTION, ENRICHMENT, STRATIFICATION, AND SAFETY MONITORING
Structural MRI
Structural MRI is currently used in HD trials for participant selection, safety monitoring and enrichment, with theoretical potential for stratification, as outlined below.
Visual expert read of structural MR scans acquired during the screening or baseline assessment (namely T1-weighted, T2-weighted, FLAIR, T2*/susceptibility weighted imaging (SWI)), is routinely performed in HD trials to exclude participants where there is evidence of a central nervous system (CNS) disorder other than HD.
Regional brain volumes, derived from T1-weighted MRI, are now used in the inclusion criteria for some HD trials. For example, in UniQure’s gene therapy study, where a micro-catheter delivers the therapy into target brain regions of the caudate and putamen, minimum volumetric values for these regions form part of the inclusion criteria for the study (www.uniqure.com/programs-pipeline/phase-1-2-clinical-trial-of-amt-130). Ensuring these brain regions are of a ‘suitable’ size, and not too atrophic, may help to ensure safe and effective surgery [31]. Here, volumetric MRI measurements aid participant selection and aim to provide biological enrichment. However, currently there is no consensus regarding safe volume estimates for surgery. Furthermore, the downside of such an approach is that many symptomatic patients may become ineligible for experimental therapy, if the volume cut-offs have not been established empirically to ensure they are appropriate for the target population. This is confounded by the large between-patient variability of regional volumetry across the clinical spectrum of HD.
Another example of where volumetric measurements will inform participant selection, particularly in preventative trials, is via the new Integrated Staging System for HD (HD-ISS) [32]. The HD-ISS uses a range of data to stage individuals through the temporal course of HD, including volumetric MRI measurements, as well as clinical and functional scores. The volumetric MRI measurements of caudate and putamen volume capture the transition into the process of neurodegeneration – reaching a certain volumetric cut-off corresponds to the participant transitioning from HD-ISS Stage 0 to Stage 1. These regions have been chosen because they provide a robust representation of the earliest sites of neurodegeneration in HD that can be reliably measured with current technology [33 –40]. With HD trials moving to earlier stages, that is preventative trials, volumetric measurements of caudate and putamen are likely to be used more widely to inform selection via the HD-ISS.
When generating regional brain volumes, careful consideration should be given to the image-analysis software selected, as well as the version number used. These can have an important impact on the volumes generated and hence the clinical decision-making, such as whether a participant fulfils inclusion criteria for a trial. For example, when calculating the HD-ISS stage using the online tool (https://enroll-hd.org/calc/html_basic.htm), Freesurfer version 6 must be used to generate caudate, putamen and intracranial volumes, since this was what was used to develop the HD-ISS cut-offs [32]. If not, the derived HD-ISS stage may be incorrect (see Knight et al., for a detailed discussion [41]). Since accurate putamen segmentation can be challenging (author experience and [42]), future refinements of the HD-ISS may include alternative software packages and associated cut-offs for the relevant package. Researchers should take care to use the same software type and version number that has been used to generate the HD-ISS cut-offs.
Particular care should be taken when using techniques that have been developed using healthy brains, since they can introduce systematic bias when applied to atrophic brains. When measuring regional brain volumes for purposes other than the HD-ISS (e.g., when using caudate atrophy as a secondary endpoint), it is a good approach to use an image-analysis technique that has already been validated within observational studies of HD, such as the longitudinal TRACK-HD study, which used the Boundary Shift Integral approach [33 –36]. Choice of image-analysis software type, version and any non-default parameters should be defined prior to the start of the trial.
Quality control of processed data is also essential, particularly where fully-automated analysis tools have been used, such as Freesurfer (https://surfer.nmr.mgh.harvard.edu/). Occasionally, an automated pipeline will fail dramatically. This is often caused by poor tissue contrast at the boundary of the structure. This can be particularly problematic for the putamen, where the continuous grey/white matter boundary can be poorly defined on T1-weighted MRI. Segmentation of the caudate is usually more accurate because the boundary of the caudate with the lateral ventricles is well defined [42, 43]. Where it is obvious that the segmentation has gross errors, the volume generated will not be reliable and the datapoint should be excluded from further analysis. However, often automated software may do a ‘reasonable’ but not optimal job. This situation is harder to manage consistently. It is essential that a standard quality control procedure is developed to deal with these cases in a consistent and scientifically robust manner. Ideally this should be defined prior to the start of the trial. Using sub-optimal software will lead to erroneous volume and volume change estimates.
In terms of participant stratification for HD trials, modelling of observational data suggests that regional volumetric measurements may be useful. In fact, including stratified randomization coupled with covariate adjustment may be beneficial, particularly for future trials with caudate atrophy as the primary outcome [44]. For example, required sample sizes for a 2-year trial with 6 monthly interim visits are estimated to be smallest if caudate atrophy is expressed as a percentage of baseline volume; stratifying by and adjusting for age, CAG repeat length, disease burden and baseline caudate volume would reduce the required sample size by 42% [44]. However, we are unaware of any trials to have adopted this approach to date, and imaging measurements are unlikely to provide primary outcomes in the foreseeable future, until they are fully validated for this use. However, this approach may prove useful in the future.
Structural MR scans are routinely used in HD trials as part of safety monitoring. Typically, T1-weighted and T2-weighted MRI are visually reviewed for abnormalities by expert neuroradiologists as part of the MRI safety evaluation process (Table 1). This process may be repeated at intervals throughout the study, as defined in the clinical protocol. Having a blinded unbiased central reader providing a second opinion to local radiological reads may safeguard against unintended confirmational bias.
Diffusion MRI
No HD clinical trials registered to date have used DWI for patient selection or stratification, although observational diffusion studies have shown sensitivity to pathological change in preHD gene carriers as they approach motor onset, suggesting DWI may have utility in identifying participants who are most likely to progress over the time-period of a clinical trial. TBSS [17] analysis in a small sample of preHD from the PREDICT-HD study showed widespread cross-sectional FA reduction and increased MD across several white matter tracts, with more pronounced differences in those closest to expected disease onset according to the CAG-age product (CAP) score [45]. There was little evidence of longitudinal change in diffusion metrics in this sample and their results suggested that volumetric measures showed greater sensitivity to change over time. Shaffer et al. [46] applied tractography in a much larger sample from the PREDICT-HD study, confirming lower baseline FA and higher MD in preHD with a higher CAP score compared to those with low CAP. Moreover, they demonstrated significantly higher rates of decline in FA and increases in MD associated with higher CAP scores, particularly in the connections between the premotor area and the putamen.
Casella et al. [15] used multi-shell DWI data [acquired with strong gradients (300 mT/m)] to study white matter microstructural differences in preHD and found higher restricted diffusion signal fraction (FR) from CHARMED in the cortico-spinal tract and reduced myelin-sensitive magnetization transfer ratio (MTR) in the posterior corpus callosum. The same pattern of larger FR and reduced myelin estimates (macromolecular proton fraction from quantitative MT) was observed in R6/1 HD mice and linked to thinner axons detected with microscopy (Casella et al., preprint https://doi.org/10.1101/2023.10.02.560424). Furthermore, preHD individuals’ CAG repeat lengths were positively correlated with MTR measurements. These results suggest that FR from CHARMED holds the potential of a cross-species MRI biomarker of axonal changes in HD that may be directly linked to the disease mutation.
Graph theory evaluates the strength of connections between brain regions or hubs, and analysis of DWI data from the TRACK-HD and TrackOn studies showed different connections were vulnerable at different disease stages [47]. For example, longer connections appeared to be affected earlier on in the disease process, suggesting this technique may provide information for staging. Nevertheless, HD-ISS, the most recent HD staging system [32] opted for volumetric biomarkers to determine cut-offs and it remains to be seen whether diffusion metrices can provide independent additional information beyond volumetric analysis.
The sensitivity of diffusion imaging to inflammation in cerebral amyloid angiopathy [48] suggests it may provide useful information for safety monitoring. Currently, DWI and the apparent diffusion coefficient (ADC; a measure of the magnitude of diffusion of water molecules in the brain), are often included as additional safety MRI sequences in HD gene-therapy trials, as they can add to the characterization of oedema following intraparenchymal injections and allow visualization of potential diffusivity restriction. Typically, the DWI and ADC map are checked for evidence of diffusivity restriction if the core safety scans indicate concerning findings [49].
Functional MRI
To the best of our knowledge, only two studies have examined the use of fMRI for patient selection and stratification in HD clinical trials [50, 51], and there is no research on the utility of ASL in clinical trials. Mason et al. [51] identified a polymarker comprised of both structural and resting-state fMRI metrics that identified premanifest HD gene-carriers with high probability of conversion within 5 years. In addition, using functional connectivity maps generated from resting-state fMRI data, Polosecki et al. [50] identified a set of features that could stratify premanifest participants into slow or fast progressing. There is therefore some early evidence that fMRI could improve patient selection in trials aiming to recruit premanifest HD patients, by identifying those with high probability of progression, but that work needs to be replicated. Furthermore, deploying such methods in a clinical trial setting would be complicated. Given the sensitivity of other methods that are easier to implement in a clinical trial setting, such as the prognostic index [52], and the introduction of the HD-ISS [32], it is unclear what is the added value of such methods for disease-modifying therapies. They could however be useful in symptomatic treatments, targeting, e.g., cognitive or psychiatric dysfunction, where there are currently no known stratificationmarkers.
MRS
To our knowledge, MRS has not been used for patient selection, enrichment, stratification, or safety monitoring purposes in HD clinical trials.
PET
A more precise prediction of the timing of phenoconversion from premanifest to manifest HD could improve patient selection and enrichment in clinical trials. As CAG repeat length and age only predict 70% of the variability in phenoconversion [53], many studies have focused on the correlation of PET imaging with phenoconversion in order to look for better prediction models.
Several [18F]fluorodeoxyglucose (FDG) PET studies, using clinical phenoconversion during follow up as reference, showed that in premanifest carriers both caudate and putaminal hypometabolism was associated with risk of phenoconversion [53, 54]. One study reported that a baseline caudate-to-pons ratio below a receiver operating characteristic (ROC)-based cut-off of 1.05 was associated with a significantly higher risk of phenoconversion within 5 years [55]. Another study concluded that normal putaminal metabolism may predict that motor symptom onset will not occur within the next 8 years [56]. Putaminal glucose metabolism in combination with psychomotor speed testing may explain two thirds of the variance in a model predicting motor onset within 5 years [56].
One longitudinal [18F]FDG PET study showed that a spatial covariance pattern of progressive striatal and cortical hypometabolism and pontocerebellar hypermetabolism was associated with disease progression on follow-up [57]. Expression of this pattern showed a significant linear relationship with time closer to phenoconversion, which gives the network increased reliability as a progression marker when closer to phenoconversion [57].
Other studies investigated the correlation of PET imaging with the Langbehn model, which predicts the 5 or 10-year probability of phenoconversion in premanifest carriers [59]. For [18F]FDG and [11C] raclopride D2/3 receptor PET, no correlations were found [60, 61]. On the other hand, the ratio of thalamic to striatal phosphodiesterase (PDE10A) binding, using [11C]IMA107, correlated with probability of phenoconversion [62]. PDE10A binding may be one of the best prognostic markers for HD motor phenoconversion [63, 64]. Also, a strong correlation has been shown between striatal TSPO binding as a marker for activated microglia/neuroinflammation and risk of phenoconversion within 5 years [61, 65] and, therefore, this may be worthy of consideration for patient stratification for disease modification trials.
An association of some PET tracers with CAG repeat length, CAP score and/or disease burden has been shown [64 , 67], but this is of limited use for clinical trials.
To our knowledge, PET imaging has not been used yet for patient selection or stratification in clinical trials in HD. However, it is a promising tool in terms of optimizing the prediction of phenoconversion in HD. As all above tracers can be quantified reliably using static quantitative protocols with simplified reference tissue approaches that do not necessitate invasive dynamic acquisitions with blood sampling, and furthermore more convenient 18F-labeled probes are available for each of the above targets (D2, TSPO, PDE10A), PET imaging should be considered for selection of appropriate HD mutation carriers for clinical trials.
MEG
To date, no specific MEG signatures for HD, nor for its specific stages, have been identified. Therefore, based on currently available evidence, we are unable to recommend its application in patient selection, stratification, enrichment, or safety monitoring for clinical trials.
SECTION 2: NEUROIMAGING AS A TOOL TO DEMONSTRATE BIODISTRIBUTION, TARGET ENGAGEMENT AND PHARMACODYNAMICS
Structural MRI
Structural MRI has limited utility for demonstrating biodistribution, target engagement or pharmacodynamics. However, for gene therapies administered to target brain regions by a micro-catheter, structural MRI is critical for selection of patients with sufficiently intact regional brain volumes, necessary for adequate local volume of distribution of the adeno-associated virus (AAV) load, delivered under convection enhanced delivery; Section 1. Structural MRI is also used for planning the trajectory of the surgery in advance. Intra-operative “real-time” MRI allows close monitoring of positioning and placement of the micro-catheter prior to the injection. Contrast-enhanced intra-operative MRI enables monitoring of the short-term biodistribution of the therapy [68].
Diffusion MRI
Previous work suggests that white matter defects measured by DWI may reflect eventual anatomical axonal degeneration. This may be important for experimental therapeutic agents that rely on anterograde or retrograde axonal transport. Besides that, with cumulative WM degradation over the course of the disease, transneuronal diffusion of mutant huntingtin (mHTT) [10] may be abnormal, and therefore it is feasible that the biodistribution of a mHTT lowering agent might be dissociated from mHTT localization in the neurons. However, further studies to establish this connection between mHTT distribution and DWI white matter metrices are required, ideally as correlational observational analysis between diffusion imaging and mHTT PET, once validated (see below). The low temporal resolution of DWI also suggests that it may not be the most sensitive tool for pharmacodynamic studies (but may be sufficient for antisense oligonucleotides (ASOs) and siRNA which have very long t
Functional MRI
Both fMRI and ASL can be used to indirectly and non-invasively measure changes in the brain induced by a pharmaceutical agent. This approach, called pharmacological or pharmaco-MRI (phMRI) [69], is applied in drug challenge trials to monitor alterations to neurotransmitters induced by drug-related changes to neurovascular coupling with a time-resolution of seconds, depending on the pharmacokinetic/pharmacodynamic (PK/PD) properties of the drug [70, 71]. Because it is non-invasive, phMRI can collect multiple measurements over short intervals to characterize signal change. Compared to PET, phMRI has many advantages for PK/PD monitoring, in terms of cost, patient burden, spatial and temporal resolution. An important difference is that the phMRI signal is indirect and non-specific, i.e., it can be used for drugs targeting many different pathways, whereas PET ligands are used for specific pathways. For this reason, detailed modelling of the drug-induced changes to neurovascular coupling and the relationship with the MRI signal is important in order to fully characterize the changes and disentangle the vascular from the therapeutic effects [70]. At present, as far as we are aware, there are no therapeutic agents in development that may likely alter CBF in HD mutation carriers. One main limitation of functional MRI methods is the limited reproducibility of outcomes [72] and therefore the statistical inferences drawn from those. While these issues are being debated, this approach has so far been used to study drugs targeting different neurotransmitters such as dopamine [73], serotonin [74] and glutamate [75].
In HD, the proximate cause of the disease pathology is production and aggregation of mHTT, which leads to multiple downstream effects including metabolic and synaptic dysfunction, axonal transport and cell survival [76]. Current disease-modifying treatments are directly targeting the cause of the disease, i.e., the production of the huntingtin protein (HTT), and if successful in halting disease progression, would also affect downstream events. These downstream effects can also lead to neurotransmitter imbalances including of glutamate [77], which could be measured using fMRI, e.g., in the form of hyperactivity. However, because of the complexity of the effects produced by mHTT and the indirect nature of the phMRI signal, if fMRI is to be used for PK/PD, detailed models are first needed describing how the effects of mHTT on different metabolic and synaptic functions relate to changes in the MRI signal, in addition to modelling the drug effects on neurovascular coupling. It is our opinion that these methods are far from being realistic or useful at the time of writing.
PET
Currently, there is a focus on developing radiotracers able to target aggregated mHTT [78]. Such radiotracers would have huge potential for the evaluation of cerebral target engagement in mHTT-lowering clinical trials; however, full validation is required before they can be adopted. To date, several preclinical studies with different PET radioligands targeting mHTT aggregates have shown promising results [78 –80]. Clinical studies with different mHTT radioligands are currently ongoing [81]. To date, only data from the [11C]CHDI-180R radioligand has been reported, and only in human healthy controls [82]. This study showed [11C]CHDI-180R is safe for use in human brain PET imaging and suggested favorable brain kinetics in healthy controls [82].
The enzyme PDE10A may be directly linked to the molecular pathophysiology of HD. Striatal PDE10A inhibitors showed promising results in a HD mouse model suggesting neuroprotective effects [83 –86]. However, a randomized controlled trial with the PDE10A inhibitor PF-02545920 (AMARYLLIS trial) in 272 HD patients failed to show efficacy in its primary and secondary clinical endpoints [87]. None of the preclinical and clinical studies with PDE10A inhibitors used PDE10A PET to evaluate target engagement or pharmacodynamics [84].
Anti-inflammatory drugs, for example non-steroidal anti-inflammatories, tumor necrosis factor-α inhibitors, monoclonal antibodies or laquinimod, an inhibitor of microglial activation, have been tested in preclinical studies and may be beneficial, with conflicting results [88]. Minocycline attenuates microglial activation, but clinical trials in human HD mutation carriers showed inconsistent results [89]. TSPO PET imaging is a promising tool to assess anti-inflammatory effects of these drugs on microglial activation [90]. A recent placebo-controlled randomized clinical trial with laquinimod in HD (LEGATO-HD) could not show any significant change of clinical manifestations or TSPO binding after treatment [91].
PET with [11C]UCB-J, a radioligand for the presynaptic terminal marker SV2A, revealed synapse loss in HD that was restricted to caudate and putamen in premanifest mutation carriers and more widespread in early manifest patients [92]. A recent study showed that corticostriatal synapses in HD mice are selectively eliminated at an early stage via a mechanism mediated by complement proteins and microglia [93]. Interestingly, administration of an antibody that blocks complement component C1q prevented cortico-striatal synapse loss in HD mice [93]. [11C]UCB-J PET would be a suitable tool to assess the effectiveness of treatments designed to rescue synapse loss in people with HD.
Cannabinoid (CB) receptor agonists could potentially have symptomatic and/or neuroprotective effects [94 –96]. CB1 receptor imaging [67] could be helpful to assess target engagement and pharmacodynamics in future clinical trials testing CB receptor agonists.
Additionally, newer PET radioligands targeting glutamate receptors might also be promising given the role of glutamatergic excitotoxicity in HD pathophysiology [97, 98].
MRS
MRS metabolites have been studied as potential biomarkers of disease progression, not as direct biomarkers of biodistribution, target engagement, or pharmacodynamics. However, as a non-invasive imaging method that enables the assessment of cerebral metabolism, MRS could be a valuable tool in the evaluation of responses to treatment.
MEG
MEG, being a functional neuroimaging technique of magnetic fields, does not directly visualize chemical compounds or structures. However, as it captures neuronal activity with millisecond temporal resolution, it has been used to demonstrate experimental modulation of neurotransmission by pharmacological agents in the studies known as “pharmaco-MEG” [99]. Neurotransmitter systems examined by pharmaco-MEG include glutamate, gamma-aminobutyric acid (GABA), acetylcholine, dopamine, serotonin, alcohol, and others (for a systematic review see [99]). Furthermore, MEG has been used to interrogate the effect of pharmacological agents on the behavior of specific neuronal populations. The approach uses dynamic causal modelling (DCM) which involves construction of a neuronal mass model that models the interaction of different neuronal populations, which is combined with a forward model describing the translation of synaptic activity into MEG signals. In a study investigating the effect of levodopa on working memory, the authors concluded that the improved working memory following levodopa administration is explained by the altered connectivity from pyramidal to stellate neurons in the superior frontal cortex [100]. In another study investigating the effect of ketamine on cortico-cortical and cortico-thalamo circuitry, DCM was successfully employed to demonstrate that ketamine increases N-methyl-D-aspartate (NMDA) and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-mediated in the superficial layer (superficial pyramidal to superficial interneuron) and NMDA and GABAA mediated self-gain of deep pyramidal neurons [101]. In a proof-of-concept study, Adams et al. performed a randomized placebo-controlled cross-over trial to assess the effect of GABA-reuptake inhibitor tiagabine on the fronto-temporal circuitry in fronto-temporal lobar degeneration [102]. The study demonstrated deficits in fronto-temporal connectivity which was related to GABA deficiency, and the biophysical model was validated by parametric Bayes analysis showing relativity between the level of GABA, measured by spectroscopy, and the posterior estimates of patients’ GABAergic synaptic connectivity. This study demonstrated the ability of DCM in pharmaco-MEG to help reveal the pathophysiology underlying neurodegenerative process, and thus potential therapeutic target.
SECTION 3: NEUROIMAGING AS A TOOL TO PROVIDE EVIDENCE FOR DISEASE MODIFICATION
In order for a neuroimaging measure to be used as evidence for disease modification it needs to be a potential or validated surrogate endpoint biomarker [103]. This means that it needs to be a reliable and sensitive marker of disease progression in HD.
Structural MRI
Volumetric MRI-based biomarkers, such as change in volume of the caudate, putamen, whole brain and CSF, are commonly included in HD trials as secondary and/or exploratory outcome measures. These volumetric readouts can be measured reproducibly, even in large multi-site studies, and are highly sensitive to disease progression during the natural course of HD [34–36 , 104–106]. Hence, they may prove useful in assessing treatment effects on brain atrophy, and ultimately providing supportive evidence for disease modification.
The Biomarker Working Group of the HD Regulatory Scientific Consortium has recently published an evidentiary review of volumetric MRI-based biomarkers for HD [107]. This review provides an excellent summary of the weight of evidence supporting the use of volumetric MRI-based biomarkers in HD trials. To summarize, regional brain volumes are reduced in HD prior to clinical motor onset; they are significantly associated with motor and cognitive function; they predict subsequent clinical progression, and they have robust longitudinal change characteristics [107]. In particular, caudate and putamen atrophy show relatively large longitudinal effect sizes and are effective several years prior to clinical motor onset [36, 39]. Global measures, such as whole-brain atrophy and CSF expansion, lack sensitivity to longitudinal change far from clinical motor onset, but are sensitive close to motor onset and in early manifest disease. They have the advantage of encompassing the global effects of neurodegeneration in a single measure. Given the uncertainty in predicting the location of therapeutic effects in the brain, and the differences in sensitivity between regions, both local measures (e.g., caudate and putamen) and global measures (e.g., whole brain, CSF space) are often included as complementary secondary and exploratory outcomes in HD clinical trials.
Interpretation of volumetric MRI-based measures could be improved with a more complete understanding of how longitudinal change in volumetric MRI-based measures relates to change in clinical readouts, on the timescale of a typical clinical trial [107]. To date, this has proved challenging, partly because the sensitivity of volumetric MRI-based biomarkers exceeds that of standard clinical instruments, particularly in premanifest and early manifest HD [104]. Additionally, we know from natural history studies that there is a relationship between change in TFC and change in volume of the caudate, whole brain, grey matter, and white matter over two years [35]. The same study showed evidence of a relationship between change in TMS and change in volume of the whole brain, putamen, grey matter, and white matter [35]. However, when there is an intervention, these relationships are more difficult to understand because of potential treatment-related side-effects, such as ventriculomegaly and inflammation (see Section 5 for a more detailed discussion).
As HD trials move earlier in the course of the disease, to preventative trials, it will be vital that we have appropriate tools to assess treatment effects on the brain. The HD Young Adult Study [108], an observational study researching individuals with the HD gene who are on average over 20 years prior to predicted clinical onset, is well positioned to inform such trials. Cross-sectional results show significantly smaller putamen volume over 20 years prior to predicted clinical onset [108]. Longitudinal findings, due in 2024, will show whether changes in the putamen (and other brain regions) can be reliably tracked longitudinally. If so, they may provide valuable outcome measures for future preventative trials.
It is worth highlighting that although volumetric MRI-based measures have been included as secondary and exploratory outcomes in many HD trials, they do not currently have regulatory qualification for this purpose. This is not a prerequisite, but it does signal a degree of evidentiary maturity and confidence in the interpretation of these measures [5]. However, many years of research has provided significant evidence on the role of caudate volume, in particular, as a sensitive biomarker in HD. Hence, it could qualify as a surrogate endpoint “reasonably likely to predict clinical benefit” in FDA accelerated approval submissions [109]; for example, in the scenario where a disease-modifying drug showed significant dose-dependent slowing of caudate atrophy combined with lowering of mHTT protein or other biomarkers. However, drug delivery and treatment-related side effects can confound efficacy effects on regional brain volumes. Further discussions on the complexities of interpreting MRI readouts in clinical trials can be found in Section 5.
Also, there is some evidence of increased perivascular space (PVS) burden over the grey matter in early HD, compared with age-matched controls [110]; although PVSs over the white matter appear relatively unaffected [111]. Further work is required to understand if changes in the grey matter are progressive. PVS expansion may prove to be an important measure, since expanded PVS may affect the distribution and success of treatments administered either intrathecally, such as nucleotide-based therapeutics, or by intraparenchymal administration of cell and viral vector-based gene therapies.
Diffusion MRI
It is well established that diffusion imaging can highlight disease-related reductions in white matter organization in both manifest [112 –115] and premanifest stages [112–114 , 116]. In the premanifest stage, increases in MD and decreases in FA are most prominent in the connections between the striatum and the sensorimotor regions [114], with MD apparently affected earliest [117]. By the time disease is manifest, diffusivity changes are widespread and involve striatal connections to the prefrontal, motor, and parietal areas [114]. More recently, NODDI and CHARMED analyses found estimates of axon density (FR, intracellular volume fraction from NODDI) were more sensitive to early pathology than standard diffusion metrics such as MD and FA in both premanifest HD [15, 16] and Alzheimer’s disease [118].
There is inconsistency in the literature in terms of longitudinal change using diffusion imaging, which may in part reflect differences in acquisition and analyses methodologies. Whilst some region-of-interest studies failed to detect longitudinal change in early HD [117, 119], others do report significant change over time in manifest disease [120, 121]. There is less evidence for longitudinal sensitivity in the premanifest phase [106, 117]. Using tractography, Harrington et al. [45] reported change in just 1 of 16 tracts examined in a cohort of premanifest HD, although a later examination of a larger sample from the PREDICT-HD study did find more widespread change [46]. It is possible that multicompartmental models such as NODDI and CHARMED may show increased sensitivity to change but longitudinal studies are currently lacking.
DWI-based models such as DTI, NODDI, and CHARMED have been developed to characterize the microstructure of white matter axonal bundles rather than the microstructural complexity of grey matter. Thus, in contrast to diffusion analysis of white matter, diffusivity changes in grey matter have shown conflicting results. Since metrics such as FA and MD are thought to reflect organization within a white-matter structure, interpretation of such changes within grey matter is more difficult. Studies have reported counter-intuitive increases in both MD and FA [115, 116], suggesting increased organization despite cell loss. Consequently, it is not clear what change in grey matter diffusion metrics in response to therapy would be expected to demonstrate effective disease modification. New approaches that exploit multi-shell DWI data to model grey matter microstructure, such as the Soma and Neurite Density Imaging (SANDI) model [122] may prove to be more suitable to probe microstructural grey matter changes.
Identifying the earliest detectable change is key as the HD field moves towards primary prevention trials. It is not known exactly when diffusivity changes become apparent, and so might be used as a disease-modification marker in the premanifest phase, although a recent study using a novel fixel-based analysis of diffusion data suggested that reductions in white-matter density were evident around 11 years before expected symptom onset. However, these changes were not detectable in an earlier cohort more than 20 years from disease onset [123]. Likewise, NODDI metrics were not sensitive at this early stage [108] but did show sensitivity in a premanifest cohort up to 15 years before onset [16].
It is important to establish the relationship between brain imaging metrics and behavioral performance if therapeutic targeting of brain structures is to translate to symptomatic benefit. Disease-related diffusion changes do correlate with both motor and cognitive function [112 , 125] and importantly a recent paper showed a close association with the composite UHDRS [126], which is currently being used as an outcome measure in HD clinical trials.
There is considerable evidence from observational studies that diffusion imaging may be a useful marker of disease modification in clinical trials. Analysis of the PADDINGTON study (61 early stage HD patients and 40 healthy controls, prospectively scanned at four sites) showed the feasibility of analyzing diffusion metrics from multi-site data; statistical evaluation of the coefficient of variation in a priori defined ROIs showed no differences between sites above chance, indicating that data were not systematically biased by center-specific factors [127]. A direct comparison of effect sizes in the same study (104) found that although FA and MD showed reduction and elevation respectively in the striatum in early HD, they would require larger sample sizes for clinical trials than volumetric measures. However, there have been improvements in both acquisition and analysis of diffusion data in recent years. Furthermore, the choice of biomarker will depend on the therapeutic target, and it is possible that diffusion imaging may prove most useful in assessing treatments designed to specifically restore white matter connections. Low dose treatment with laquinimod appears to improve white-matter microstructural abnormalities in the YAC128 mouse model of HD (116), demonstrating its potential for future clinical trials.
Functional MRI
At present there are no fMRI or ASL metrics that could fulfil the criteria of a surrogate biomarker in HD. However, multiple studies have described disease-related changes in brain function in HD, which can be used as early exploratory endpoints to understand whether treatment can help restore function. Caution should be exercised however not to over-interpret or over-generalize such findings as they have not been studied extensively.
Cross-sectional fMRI studies in HD have reported a complex pattern of differences compared to controls including both increases and decreases in brain activity and connectivity [128 –132], which suggests that fMRI captures both pathological processes and compensatory mechanisms. Longitudinally, differences in the rate of change between HD gene-carriers and controls and correlation between rate of change and clinical measures have only been reported using task-fMRI [125 , 134], but not resting-stage fMRI [135 –137]. Using a shifting response set task, the IMAGE-HD [138] study showed that brain activity in frontal regions, the striatum and the cingulate, decreased in symptomatic HD participants compared to controls and correlated with clinical dysfunction [125] over 30 months. Such a pattern of change is consistent with the presence of neurodegeneration and suggests that task-fMRI may be sensitive to disease progression. However, using an n-back task [139] the same study showed increase in fMRI activity in premanifest HD participants compared to controls over 18 and 30 months in the frontal lobe, the striatum and the cingulate [134]. The increase over time in fMRI activity in the frontal lobe and the striatum was higher in premanifest HD gene-carriers further from disease onset and lower in those closer to disease onset [134]. This pattern of change could be a result of both pathological and compensatory processes (or attempted compensation) in premanifest HD [129, 132]. Successful treatment with a disease-modifying therapy could normalize pathological brain activity and slow neurodegeneration. phMRI could therefore be used to test whether treatment normalizes brain activity, e.g., reduces regional hyperactivity or hypoactivity [140], by comparing brain activity before and after initiation of treatment.
For ASL there is only a handful of small studies in HD [141 –143] which show differences in perfusion between HD gene-carriers and controls in many different regions, but none are longitudinal. It is therefore unclear whether ASL can be used to monitor changes following disease-modifying therapies and what would be a suitable endpoint.
MRS
Alterations of NAA or total NAA (tNAA), MI and total creatine have been reported in HD gene expansion carriers, e.g., [144 –146], but evidence from longitudinal studies that these (or other) MRS metabolites demonstrate change over typical clinical trial durations is still scarce. One paper reporting on longitudinal MRS data collected as part of the TRACK-HD study concluded that while putaminal NAA and tNAA were reduced and MI elevated in early HD relative to healthy controls, the group differences remained consistent over two years of follow-up [144]. Analyzing baseline and 2-year follow-up MRS data from the HD-CSF study of gene expansion carriers and healthy controls, a more recent study reported no significant change in putaminal total creatine and MI concentrations in gene expansion carriers over the follow-up period [146].
Correlations of metabolites including NAA/tNAA have been reported with both disease burden score [144] and motor/visual/cognitive performance [145], signaling the potential utility of MRS metabolites in providing supportive evidence of therapeutic responses. However, findings from different studies with varying data acquisition, analysis, and quality control protocols have not always been in agreement [146]. The HD-CSF cohort study evaluating putaminal metabolic profiles also analyzed volumetric imaging data and found significant associations of caudate volume with total creatine and MI, at both timepoints. However, the metabolite concentrations showed no significant groupwise differences between “premanifest” and “manifest” gene expansion carriers (stratified by Diagnostic Confidence Level and CAG repeat length) when age and CAG repeat length were controlled for. The same study found relationships between MRS metabolites and biofluid markers, including a positive correlation between MI and plasma Neurofilament light (NfL) protein at baseline and inverse correlations of total creatine with mHTT, CSF tau, CSF NfL and plasma NfL at the 2-year follow up [146]. Such relationships are interesting as MRS can offer a non-invasive way of quantifying biochemical alterations reflective of neuronal/axonal damage.
Overall, studies have reported abnormalities of MRS metabolites in gene expansion carriers relative to healthy controls (especially reduced putaminal/striatal NAA/tNAA), as well as correlations with select clinical, imaging and biofluid markers known to change as HD progresses. However, evidence in support of MRS as a source of HD biomarkers of disease progression/treatment response remains inconclusive. Future work should include multi-site assessments of longitudinal datasets from a clinically and neuropathogically diverse HD population, in addition to consideration as to which MRS-based metabolomics and metabonomics might best illuminate complex interactions of various neurochemicals and metabolites under progressing pathological conditions in HD populations [147].
PET
PET imaging could be useful as a surrogate quantitative marker of motor or cognitive symptoms, as the use of clinical scales has its limitations. Also, in the premanifest stage, where cognitive or psychiatric symptoms can be very subtle and difficult to detect or follow clinically [35], PET imaging may be of added value.
First, many studies in this domain have been performed with [18F]FDG PET. A trend for a correlation between basal ganglia hypometabolism and slower psychomotor speed was found in premanifest carriers cross-sectionally [56]. Also, striatal and cortical hypometabolism were associated with functional capacity and motor function [148] or verbal learning, memory and cognitive impairment [149, 150] in manifest HD patients. However, another study failed to show such correlation [66]. A recent study demonstrated cortical hypometabolism in premanifest and manifest HD was related to arithmetic word-problem solving difficulties [151]. Apathy was associated with cortical and limbic metabolic network changes in early manifest HD [152]. Second, decreased striatal D2 receptor binding in premanifest carriers was correlated with executive dysfunction and lower verbal fluency [153]. Whilst another study showed that decreased hypothalamic D2 receptors may be associated with some prodromal symptoms [154]. In manifest HD, D2 receptor loss correlated with motor impairment [155, 156] and functional capacity [156]. Also, frontotemporal D1 receptor loss correlated with cognitive performance [157] and the presence of rigidity was associated with higher reduction in D1 and D2 receptors in manifest HD [158]. Furthermore, in a cross-sectional study with premanifest and manifest HD carriers, loss of striatal PDE10A binding was strongly correlated with motor impairment [159], however, in early premanifest carriers, extrastriatal PDE10A loss was not associated with clinical scores [160]. Also, striatal TSPO binding correlated positively with motor impairment [61, 161] and disease burden score [61]. Total functional capacity in manifest HD showed a negative correlation with TSPO binding in certain cortical regions important for cognitive functioning [61]. Finally, loss of [11C]UCB-J binding, a marker for presynaptic terminals [162, 163], in putamen was associated with motor impairment [92]. PET imaging targeting the CB1 receptor [164], a presynaptic protein, showed that prefrontal and cingulate CB1 loss was correlated with behavioral problems in premanifest HD [165].
Taken together, motor, cognitive, and behavioral impairment showed correlations with uptake of different cerebral molecular PET targets. Nevertheless, as most of these correlations have not been widely replicated, and a causal relationship of the associations has not been proven, combining PET imaging with clinical scoring is still preferable.
For future disease-modification trials, a deceleration of progressive [18F]FDG hypometabolism or slowing of decrease of D2 receptors and PDE10A may point to a protective action of the drug under investigation. Several previous PET studies investigated changes of these imaging modalities across HD stages without interventions. First, progressive striatal and cortical glucose hypometabolism has been described, even in premanifest HD carriers far from motor symptom onset [60, 66]. One longitudinal study reported a (non-significant) mean annualized striatal decrease of 2.3% in premanifest HD [166]. In early manifest HD, larger longitudinal decrease in cortical [18F]FDG uptake compared to controls was associated with faster disease progression [167]. Second, longitudinal D2 receptor imaging studies reported mean annualized striatal loss of 4.0% to 6.3% in mutation carriers compared to controls [155 , 168]. The annual decrease was higher in caudate than in putamen. Moreover, a significant correlation between disease duration and striatal D2 receptor binding was found [155]. The mean annualized decrease rate seemed to be slightly higher (–4.0%) in premanifest than in manifest carriers (–3.0%) [156]. A longitudinal study failed to show a correlation between motor symptom progression and advancing striatal D2 receptor decrease [54, 155]. Longitudinal decrease in frontal and temporal D2 receptors was associated with decline in executive functioning and attention [169]. Another longitudinal study only showed correlations between progressive executive dysfunction and striatal D2 loss [155]. Finally, studies with PDE10A PET showed progressive striatal PDE10A loss over HD stages [54, 170]. Two longitudinal studies including premanifest and manifest HD mutation carriers showed an annual decrease of 5.9 to 16.6% in caudate nucleus, 4.4 to 6.9% in putamen and 4.3 to 5.8% in globus pallidus [63, 64]. Again, there was a trend towards faster decline in earlier stages [63, 64]. Annualized changes in PDE10A could not predict clinical progression [64]. A recent longitudinal study did not find significant changes in cortical or subcortical SV2A uptake in early HD after 2 years of follow-up [171]. Interestingly, the rate of SV2A loss tended to be higher in premanifest than in early manifest subjects, while the rate of volume loss as measured by MR showed the opposite trend [171]. Future longitudinal studies comparing [11C]UCB-J PET and volumetric MRI in larger cohorts of premanifest subjects should assess the potential value of [11C]UCB-J PET for monitoring disease progression in premanifest HD. No longitudinal TSPO or CB1 PET studies are available yet.
In summary, the rate of change with disease progression seems larger for PDE10A than for [18F]FDG and D2 receptor imaging, and [18F]FDG PET seems least sensitive for monitoring disease progression as its annualized change is smaller than for PDE10A and D2 receptor imaging. However, no longitudinal head-to-head trials comparing these three modalities are available [64, 86]. Regarding their use to monitor progression in clinical trials, [18F]FDG, D2 receptor and PDE10A PET appear to be most useful in earlier, i.e., premanifest HD stages, because the annual decrease on [18F]FDG, PDE10A, D2 receptor imaging appears to be non-linear, tapering off in later stages [64 , 172]
PET radioligands targeting mHTT aggregates would be an invaluable tool to monitor disease modification in clinical trials investigating potential new drugs, e.g., mHTT-lowering therapies [76 , 88]. Nevertheless, for mHTT PET imaging to be useful as a biomarker of disease progression, further work assessing correlations between clinical progression and progressive mHTT imaging changes, will be essential.
Several disease-modification trials investigating potential new drugs, incorporated [18F]FDG or TSPO PET imaging in the study protocol to evaluate changes in brain metabolism or inflammation, respectively. The SIGNAL randomized controlled trial evaluated safety and efficacy of anti-semaphorin antibodies in premanifest and manifest HD, which may influence chronic microglial and astrocytic activation [173, 174]. Both [18F]FDG and TSPO PET imaging were used during longitudinal follow-up of study participants. The study failed to reach two of three primary clinical endpoints, however, neuroimaging results were promising as [18F]FDG PET showed increased glucose metabolism in a majority of brain regions 17 months after start of treatment [175]. Also, a small placebo-controlled trial investigating riluzole showed a beneficial effect on cortical glucose metabolism [176]; however, a much larger study found no clinical improvement with riluzole [177]. Moreover, one trial with lamotrigine suggested less decrease of glucose metabolism in parieto-occipital cortex and cerebellum after treatment in manifest HD [178] and a small study showed increased cortical and thalamic metabolism after pridopidine treatment [179]. Finally, as described in section 2, the LEGATO-HD trial failed to show a significant effect of laquinimod treatment on TSPO-PET [91].
MEG
As outlined above, there are very few studies to date applying MEG in HD. Whilst no HD-specific MEG signatures have yet been established, it is possible that MEG may provide evidence of disease modification through maintenance of baseline measurements. Studies to characterize the MEG signals at different clinical stages of HD, together with an investigation of sensitivity to longitudinal change, are first required. However, there is some normative data and studies of other neurodegenerative conditions which suggest possible utility in HD [180 –182]. For example, a study investigating the cortico-subcortical neuronal network direction in Parkinson’s disease identified significant network differences between the patient group and healthy control group using beamformer [182]. Additionally, associations between MEG signal and clinical function (mainly cognitive decline) have also been demonstrated in Parkinson’s disease in a longitudinal study, using two resting state MEG recordings, four years apart [183]. In view of this, application of MEG at different clinical stages of HD [32] may reveal the characteristic MEG signals that correspond to each stage, at both deep striatal and superficial cortical level. These may be of use in evaluating interventions in the future.
SECTION 4: NEUROIMAGING AS A TOOL TO UNDERSTAND BRAIN RE-ORGANIZATION FOLLOWING THERAPY
Structural MRI
Structural MRI has not yet been used as a tool to understand brain re-organization following therapy in HD. However, recent advances in quantitative MRI techniques, such as multi-parametric mapping (MPMs), quantitative magnetization transfer imaging (qMT) and quantitative magnetic susceptibility imaging (QSM), enable the quantification of specific microstructural brain tissue properties, such as myelin and iron content. These may prove informative in the future, particularly since it has been proposed that myelin may play an important role in the process of neuroplasticity [184, 185].
QSM data at 7T suggest altered brain iron levels in the caudate, putamen and globus pallidus in premanifest HD [186, 187] and early manifest HD [186], with some evidence of increased brain iron deposition rates over 1 year [186]. In concordance, analysis of MPMs at 3 T in a cohort of premanifest individuals ∼20 years prior to predicted clinical onset, suggests increased iron in the subcortical structures and the surrounding white matter [188]. The same study showed that reduced myelin or iron (i.e., lower R2*) was associated with higher CSF neurofilament light (NfL), a marker of axonal degeneration, in the frontal lobe and parieto-occipital cortices [188]. Macromolecular Protein Fraction (MMPF), a putative marker of myelin, derived from qMT data combined with diffusion-weighted MRI data, has shown HD-related reductions in MMPF at 3T, suggesting that myelin breakdown contributes to white matter impairment in HD [189]. Additional evidence supporting myelin breakdown as an early feature of HD was provided by Casella et al. [190] who demonstrated lower myelin water signal fraction within the callosal region in premanifest HD, using gradient echo MRI at 7T. In summary, quantitative MRI metrics have potential to inform us about disease pathogenesis in HD and they appear to be sensitive to early changes; however, much more work is required in this area to replicate and extend initial findings. Furthermore, based on human and animal studies, it has been proposed that myelin may play an important role in the process of neuroplasticity [184, 185]. Hence, it may be possible to use quantitative MRI readouts, such as those described above, as a tool to monitor structural brain reorganization following therapy in HD, via changes in myelin. Again, further work is first required to fully characterize changes in quantitative MRI metrics during the natural course of HD, prior to their application in a clinical trial. Additionally, it should be noted that the quantitative MRI acquisitions are different from the standard T1-weighted structural scans and would require additional scanning time.
Diffusion MRI
Brain plasticity in response to physical activities such as juggling [191] or balancing [192] and cognitive training in language [193] and reasoning [194] has been demonstrated using diffusion imaging. There is also diffusion-based evidence of brain reorganization in response to pathology such as glioma [195] or stroke [196]. Although there are currently no reports of brain reorganization in response to therapy in HD, one study used DWI to assess pre- and post-operative structural connectivity following deep brain stimulation in Parkinson’s disease [197] suggesting this modality may be sensitive to therapies targeting white-matter restoration or even remyelination. Furthermore, HD-DRUM, a feasibility RCT, will model white-matter (myelin) and grey-matter plasticity with CHARMED, SANDI, and qMT techniques after 2 months of rhythmic auditory stimulation training in preHD and HD (TFC > = 9) participants (Ioakeimidis et al. under review, preprint https://doi.org/10.1101/2023.11.15.23298581). Data from this trial will determine the feasibility of a larger effectiveness trial for HD-DRUM, and will also advance our understanding of the neural mechanisms underlying training effects in HD. These outcomes will inform future clinical design in this domain.
Functional MRI
In the process of restoring brain function, successful disease-modifying treatments could also enhance repair mechanisms. It is currently unclear whether such processes could be measured using MRI. FMRI has been extensively used to examine brain plasticity and re-organization in rehabilitation [198], training [199] or neurostimulation [200], as such it is well-suited for the study of restorative processes following disease-modifying therapies. Such restorative processes could involve enhancement of brain activity or connectivity [201, 202], or changes in CBF [202], as the brain normalizes its function. These changes could also mirror compensatory processes that have been observed in HD due to neurodegeneration [203], with increases and decreases of the signal over a long period of time. However, the key difference would be that these changes would be coupled with reduction in brain pathology, e.g., mHTT lowering or slower striatal atrophy, rather than with increasing brain pathology [129].
PET
The importance of structural and functional networks is increasingly recognized in HD and other neurodegenerative disorders [204 –206]. mHTT may spread trans-synaptically through neuronal networks [205, 207], which may contribute to progressive neuronal disconnection [204, 208]. Also, wild-type huntingtin is likely involved in normal synaptic function and it has been shown that synaptic dysfunction plays a substantial role in the pathophysiology of early HD [209]. PET with [11C]UCB-J, a radiotracer targeting presynaptic SV2A, can be considered a proxy for brain synaptic density. A cross-sectional [11C]UCB-J PET study in premanifest and manifest HD carriers reported cortical and subcortical loss of presynaptic terminals, indicating widespread synaptic disconnection [92].
Resting-state fMRI (rsfMRI) studies showed functional brain network re-organization even in early stages of HD [50, 210] and regional cerebral glucose metabolism was shown to be interrelated with rsfMRI [211 –213]. Also, an HD-related spatial metabolic covariance pattern, which is increasingly expressed with disease progression, has been described [57]. Given the suggested link between rsfMRI and brain metabolism, [18F]FDG PET-based metabolic covariance patterns can contribute to a better understanding of brain network re-organization. Hybrid PET/MR offer simultaneous multimodal functional imaging which may provide novel insights in brain metabolic or functional network reorganization.
MRS
To our knowledge, MRS has not yet been used in HD clinical research as a tool to understand brain re-organization following therapy.
MEG
There is an abundance of literature regarding the use of MEG for investigating neuroplasticity following stroke [214], traumatic brain injury [215], brain tumors [216], or neurosurgery [217], and also evaluating cortical functions such as aphasia [218], weakness, and visual impairment. However, most existing studies have focused on examining cortical responses and/or functions. The hallmark of HD pathology is within the deep grey-matter structures, particularly the striatum; however, application of DCM and beamformer, will theoretically enable assessment of neuroplasticity following therapy targeting the disease process in HD. Further work is required.
SECTION 5: CHALLENGES FOR TRANSLATION TO CLINICAL TRIALS
Structural MRI
How neuroimaging can facilitate clinical trials in HD
✓ ✓ Current application/adoption in HD clinical trials. ✓ Potential to facilitate clinical trials based on observational data in HD. ? Theoretical potential or potential based on data in other disorders. More information required for HD. ×Unlikely to be useful in a clinical trial setting in HD.
Logistics: Of the imaging modalities discussed in this Opinion Article, structural MRI is the most mature, already playing an important role in HD clinical trials, some with over 100 sites (e.g., LEGATO-HD study, [219]); Central imaging facilities are well-positioned to set-up the scanning at each site, collect and analyze the MR data. Harmonized acquisition sequences are readily available (e.g., https://adni.loni.usc.edu/wp-content/uploads/2017/07/ADNI3-MRI-protocols.pdf), ensuring standardization of data across sites. A high-quality 3D T1-weighted scan, used for volumetric MRI-based measures, can be acquired in 5–8 minutes, so the participant burden is relatively low (Table 3). In reality, most studies will acquire a range of MR scans during one scanning session (e.g., additional safety scans, DWI, rsfMRI, MRS) and the participant may spend around an hour in the scanner. For structural scanning, no bespoke ancillary equipment is required, making the set up straightforward. However, for reliable longitudinal data, it is imperative to have consistency of scanner hardware over time. As a minimum, study participants should be scanned on the same scanner at each timepoint. Scanner hardware and software upgrades during the study should be avoided wherever possible and managed carefully by the central imaging facility, if unavoidable. Scan acquisition parameters, and positioning of the participant with respect to the isocenter of the magnet, must also be consistent throughout the study.
Practical considerations of implementing neuroimaging into large-scale clinical trials in HD
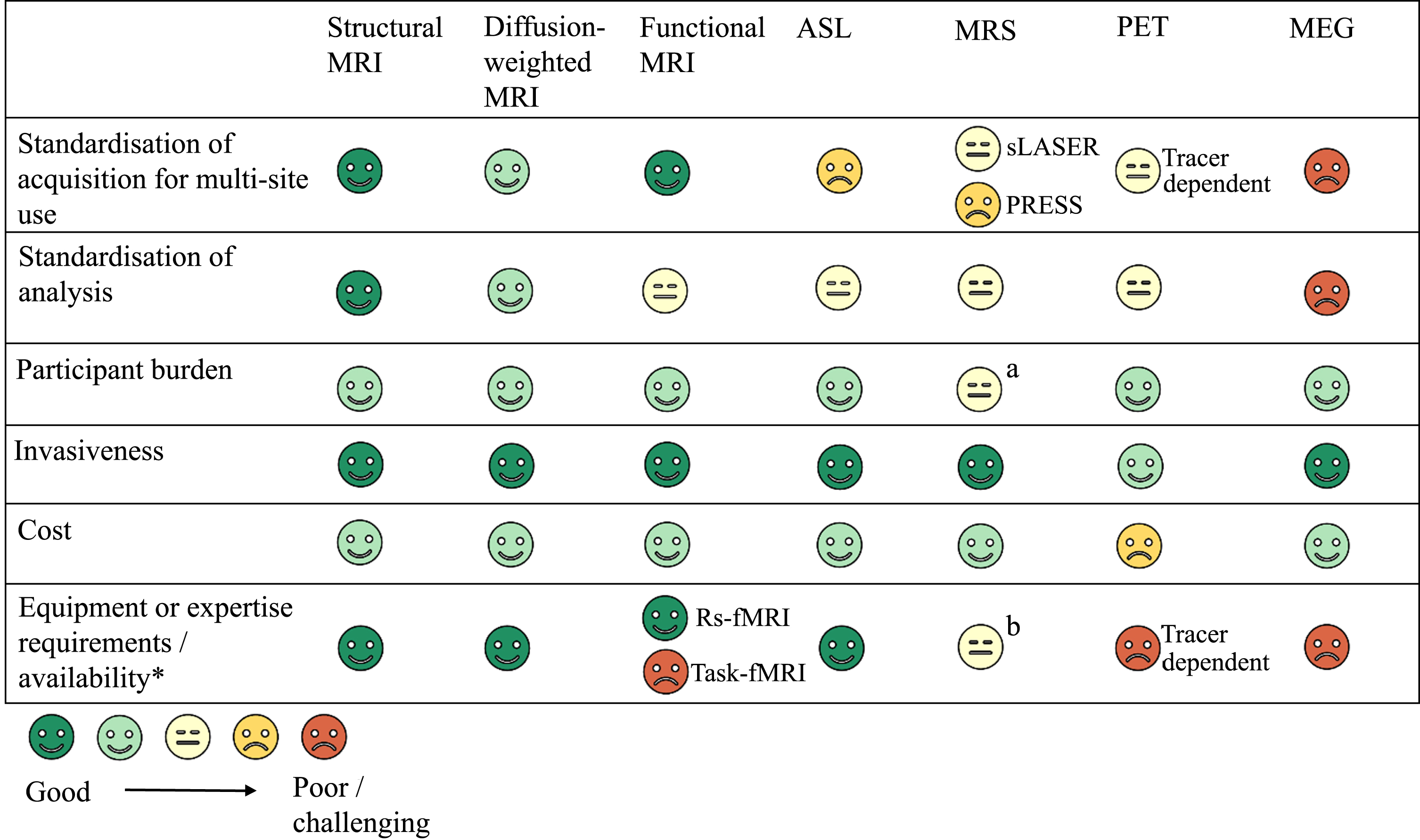
*Relative to other imaging modalities. aParticipant burden is increased relative to the other MRI techniques because voxel positioning requires extra time (5–8 mins) before the actual scan can be performed. Failed MRS acquisitions, often due to lack of MRS expertise at the site, lead to rescan requests and therefore additional study visits for the participant, particularly if the scan is for an important timepoint and the study sponsor needs usable MRS data for that visit. bNot all sites have the expertise in the radiology team to get MRS data collection right.
The cost of sMRI is reasonable compared with some other imaging modalities, such as PET. If the participant moves in the scanner, this will affect the quality of the scan and the reliability of the result. Suitable quality control should be in place to either rescan individuals where the scan shows motion artefact, or to remove them from the study. Using trained radiography staff with an understanding of HD can have a significant impact on reducing the issue of movement by, e.g., including additional padding and regular interaction during scanning, although this may not be feasible for all sites.
There are some contraindications to MRI including implanted devices that may contain metal such as pacemakers, aneurism clips, cochlear implants, and intraocular metallic foreign bodies. These aspects of MRI safety will need to be considered during screening. If MRI is a core part of the study, some participants will need to be excluded if they are not suitable for MRI. This could be particularly detrimental for intraparenchymal delivered AAV, and also in intrathecal cisterna magna/intracerebroventricular deliveries, where trajectory planning is a pre-requisite (see Section 2).
Interpretation: Reductions in global or regional brain volumes measured from MRI are typically attributed to atrophy – a loss of neurons and the connections between neurons – caused by the neurodegenerative process. However, brain volumes also include other microstructural components, such as glial and astrocyte cells, cell processes and the extracellular environment [5]. In a clinical trial, a therapy may cause changes to more than one microstructural component and hence the result can be more complex to interpret than in a natural history study. For example, if a therapy were to reduce inflammation, this may change the MRI signal and result in ‘accelerated’ atrophy readouts. This is often referred to as pseudoatrophy and is linked to the reduction in inflammation, rather than neuronal loss. Paradoxical changes in brain volume due to anti-inflammatory effects have been observed in MS trials, and are a possible explanation for puzzling brain volume readouts in anti-amyloid-β therapies in AD (see recent editorial by Barkhof and Knopman for a more detailed discussion [220]). There is also evidence that hydration can confound MRI-derived volume measurements [221]. So, if a therapeutic were to cause regional hydration changes, this might also impact the atrophy result. Some HD trials have reported substantial reductions in caudate atrophy rates in treated groups compared with placebo, e.g., 25% –50% [175 , 223]. It should be considered whether such signals have been driven by non-neuronal factors. For example, in trials with anti-inflammatory drugs like SIGNAL-HD or LEGATO-HD, ∼30% slowing of caudate atrophy was observed in the treated compared to the placebo group, but neither of the studies met their clinical endpoints. The presence of considerable slowing of caudate atrophy without a clear relationship with clinical outcomes raises questions. Is there another effect that could be confounding the signal, resulting in apparent slowing of atrophy, such as hydration changes? Alternatively, if the change measured does represent true slowing of atrophy, is it clinically meaningful? In the SIGNAL-HD study, even though the treatment group had 26% slower atrophy rate than the placebo group, it still had on average 4.4% atrophy at month 17. This could imply that either longer treatment periods are required, or this amount of slowing is not sufficient to observe clinical benefit. As an aside, the triheptanoin study [223], which showed a substantial ∼50% reduction in caudate atrophy rates in treated versus placebo groups over 12 months, used an external control group, and it is possible that other factors have driven these differences.
Another event that could lead to pseudoatrophy is large expansion of the ventricular space, such as the cases of ventriculomegaly that were reported in the Tominersen trial [224, 225]. In such cases, regional compression and displacement due the very large ventricular expansion can lead to pseudoatrophy [226]. Some algorithms are robust to the presence of regional displacement, for example, the caudate BSI [29], but it is very difficult to account for the effects of compression or transepenymal flow. However, in such cases, regional volume should return to previous levels following a reduction of ventricular volume, for example, after discontinuation of treatment.
Careful planning can help to minimize the potential impact of pseudoatrophy. For example, considering the timing of the MRI scan with respect to the treatment, and the number and frequency of imaging time-points in the study. Treatment-induced changes in inflammation, for example, may be transient, and associated changes in brain volume may be relatively short term, whereas treatments that slow the rate of neuronal death should result in a longer term and more consistent atrophy readouts. Hence the timing of the baseline scan with respect to administration of therapy is important, but also acquiring multiple longitudinal follow ups and plotting the trajectories of volume change may help to clarify what is happening (see review by Schwarz [5]). Additionally, combining the results from different imaging modalities is likely to aid interpretation (see Section 6).
Volumetric MRI-based measurements may not be suitable for assessing the efficacy of all types of intervention. For example, caudate atrophy may not be a suitable outcome measure in gene therapy trials, where the therapy is administered via stereotactic neurosurgery directly into the caudate. Currently, there is a sparsity of data on how this type of surgery may impact imaging biomarkers in HD; however, there are reports of MRI abnormalities at the site of the catheter tip (localized to the site of administration) following intraparenchymal catheter-based vector delivery in other diseases [227]. In the event that the morphology of the region is directly altered by the drug administration, the post-operative volumetric measurements may no longer follow the natural history trajectory of the disease and it would therefore be difficult to interpret or associate with clinical outcomes. To compensate, adaptations to image-analysis pipelines may be required, however because these changes are physiological (and not artifacts) they cannot be eliminated from the MRI signal and would be difficult to disentangle from other physiological processes. As these therapies advance in HD, good communication and sharing of trial findings will be critical in understanding how these techniques impact on the MRI signal and related biomarkers. Furthermore, for these trials, there may be value in including additional brain regions as biomarkers. This would enable the comparison of atrophy results generated from regions directly impacted by the administration of the gene therapy (such as the caudate, putamen) with those that are not impacted directly but still relevant to the disease (e.g., total white matter, total grey matter, CSF/lateral ventricles, whole brain).
Another important point for consideration is the distribution of molecules to the brain. Unequal distribution, such that some regions receive a higher concentration than others, could lead to differences in the rate of atrophy. This has been reported in case of intrathecally administered ASOs, e.g., Tominersen, where there was higher concentration of Tominersen in the cortex, but lower in the striatum [3, 228]. In this case, it is unclear whether endpoints like caudate volume will be useful as a measure of efficacy, even if the drug had the desired effect in other regions of the brain. Alternative endpoints, such as whole-brain or total grey matter volume, could be more informative in these cases.
In summary, when selecting volumetric MRI-based biomarkers for HD trials, it is important to consider the disease stage of the participants, the mechanism of action of the therapy, the mode of delivery of the therapy, the distribution of the molecule to the brain, as well as the timing and number of MRI visits. High-quality data acquisition is of critical importance, as is the use of suitable analysis pipelines, already validated for use in HD, combined with rigorous quality control processes.
Diffusion MRI
Diffusion imaging can easily be acquired on standard 3T scanners either in research or a clinical setting with low patient burden in terms of scanning times, so is practical for large-scale implementation. Acquisition time is of critical importance to minimize discomfort and reduce movement artefacts occurring in the signals collected from those patients with chorea. Diffusion sequences are also typically noisier and more disruptive than standard sequences so may be more distressing to patients.
This modality has good test-retest reliability [229] with longitudinal consistency [230] although this varies depending on the metric and region of interest studied. FA and MD are historically the most widely used metrics but measures from multicompartment models such as NODDI, CHARMED, and SANDI have the potential to provide more specific biological information and have been shown to demonstrate good reproducibility [231]. However, there may be a trade-off between the additional time required to acquire multi-shell data if the more advanced multicompartment models are going to be used for analysis. Many different parameters must be considered for acquisition selection. For example, in NODDI the intracellular volume fraction is most affected by the b value whereas orientation dispersion index is affected by gradient directions [232]. Ultimately, there may need to be a compromise over the optimal acquisition sequence in order to achieve cross-scanner consistency. There is also variability in the preprocessing techniques and analysis models, all of which will have a significant impact on derived metrics and their reliability. For example, a recent study by Borrelli et al. [233] showed the highest reproducibility with probabilistic constrained spherical deconvolution modelling and a high b value acquisition. Consequently, there is a pressing need to standardize acquisition, pre-processing, and image analysis techniques, so that appropriate comparisons between metrics can be made and robust reproducible tools can be implemented to generate outcome measures for clinical trials.
Functional MRI
PhMRI can play an important role in drug development in HD. As mentioned, it can be used to understand a drug’s PK/PD, as well as characterize restorative processes in the brain following a disease-modifying therapy in carefully selected experimental therapeutics. However, in order to successfully apply phMRI in clinical trials there are a few important considerations regarding trial design and selection of appropriate endpoints. First and foremost, it is important to develop models of the effects of a pharmaceutical agent on neurovascular coupling [69, 234], otherwise it will be very difficult to interpret post-treatment changes in fMRI and ASL signal.
Preclinical phMRI studies are key for this purpose and can be translated from disease models to humans. For fMRI in particular an additional consideration is which type to use, task- or resting-state fMRI. Task-fMRI is a reliable and sensitive method of probing cognitive function. In HD, working memory tasks have been used successfully to measure differences in brain activity between HD gene-carriers and controls [133 , 236] and can be used in clinical trials. A major disadvantage of task-fMRI is that it requires technician expertise to deliver the task, participant training on the task, as well as specialized stimulus delivery and recording setup, which is not readily available in many clinical sites. It is therefore not easy to deploy in large multi-center trials but could be deployed in a subset of sites with experience in collecting task-fMRI data.
Resting-state fMRI, on the other hand, does not require a specialized setup and it is therefore easy to apply in a clinical trial setup. A drawback of resting-state fMRI is that it has very low test-retest reliability [237], which reduces the ability to measure longitudinal change. To increase reliability, scan time needs to be increased significantly, which increases patient burden and study costs. An alternative approach to resting-state fMRI is to use naturalistic viewing paradigms [238], i.e., movie clips. Naturalistic viewing paradigms create a controlled environment, which increases signal reliability, without the additional burden of long scan times and the need for specialized setup and training. The only prerequisite for a naturalistic viewing paradigm is the ability to project a movie to the MRI scanner room. An additional advantage of this approach is that movie clips can be designed to allow the probing of specific cognitive functions, creating a hybrid paradigm between task- and resting-state fMRI. There are currently no published naturalistic studies in HD, despite progress in other diseases [239], but it is an area that holds a lot of promise for phMRI and drug development.
Compared to measuring brain activity and connectivity using fMRI, ASL is easier to deploy in a clinical trial setup. There is no need for specialized setup or training and the signal is highly reliable particularly for pseudo-continuous ASL (pCASL) [240]. A major disadvantage of ASL is that until recently there was no consensus regarding optimal sequence parameters [241], there is therefore large variability in methods used in the literature. In addition, pCASL product sequences are only available in GE and recent software versions in Philips and Siemens scanners.
PET
First, molecular PET imaging has some specific technical and processing challenges. PET tracer availability may be a concern depending on the site locations, especially for non-commercially available tracers or radioligands with a short half-life of the radionuclide, e.g., 11C, which need to be made on-site. However, most of the tracers reviewed here can be labeled using 18F, which can alleviate some of the distribution issues. In the past, multicenter trials with different PET scanners may have led to increased image data variability; however, current time-of-flight PET cameras have more comparable improved 3–5 mm spatial resolution, sensitivity and technical correction factors, which augments comparability. When used in a longitudinal design, test-retest variability can be challenging for some tracers with invasive quantification, but with tracers that allow reference tissue approaches, this is less of an issue. For some tracers, PET image acquisition may be influenced by confounding factors. Sensory stimulation after tracer injection, blood glucose levels or medication may affect [18F]FDG uptake. Also, neuro-inflammation in the brain may mask regional neuronal hypometabolism [242]. Cerebral TSPO binding may be influenced by infection or peripheral inflammation [243]. For specific tracers without simplified quantification or widespread distribution, dynamic PET image acquisition with arterial catheterization may be needed, which is more invasive and requires site expertise. The appropriate choice of reference region is important and can provide challenges, especially for tracers with brain-wide target distribution such as [18F]FDG or SV2A tracers [244]. An ideal reference region needs to be devoid of disease pathology and specific tracer binding, which is particularly challenging in multisystem brain disorders such as HD. In previous [18F]FDG studies, different (pseudo-)reference regions such as whole grey matter [56, 66], pons [55, 60] or cerebellum [152, 167] have been used. A recent study suggested the pons as a suitable reference region for [18F]FDG PET imaging in HD [244]. For SV2A PET, centrum semi-ovale has been validated as a suitable reference region in healthy controls as well as in different neurodegenerative disorders other than HD [245, 246]. Nevertheless, the widespread pathology in HD involving nearly all brain regions [247, 248] makes the selection of an appropriate reference region complex and challenging.
Second, molecular PET acquisition in HD mutation carriers poses some particular challenges. Imaging in manifest HD patients is prone to motion artefacts due to chorea and cognitive deficits. As the spatial resolution of PET imaging is around 3–5 mm for current systems, partial volume effects need to be considered for quantification, as the HD brain shows progressive atrophy [35], but also certain structures of interest are particularly susceptible to spill-in or spill-out of adjacent regions. For example, the caudate nucleus is situated alongside the ventricles, and the globus pallidus is bordering the putamen and close to the ventricles. Various methods for correction of partial volume effects have been suggested, so comparison of present-day and historical studies using less-elaborated partial volume correction methods may be difficult [249].
Third, translation of PET findings to clinical practice is not always straightforward. Firstly, there are few large, longitudinal follow-up PET studies in premanifest and manifest carriers, and only a small number of head-to-head trials comparing different imaging modalities. Correlations between certain PET imaging modalities and clinical scores in previous studies were not always consistent. Possible explanations for this are different acquisition protocols or data processing methods and relatively small sample sizes. Furthermore, changes in imaging measures do not always correlate with clinical benefit[35, 174].
MRS
There are some key challenges with implementing MRS in HD clinical trials. Which metabolites can be quantified as endpoints depends on factors including the pulse sequence and achievable spectral resolution and signal-to-noise ratio [250]. Advanced acquisition can improve the quality of MRS data, which results in more accurate and reproducible endpoint measurements from analysis protocols. However, MRS data acquisition normally requires local expertise, especially using advanced protocols that can be challenging to implement [251]. In trials, MRS expertise is needed both during site qualification and during the trial to avoid protocol deviations, scan failures (and therefore potential rescans), poor data quality, and inconsistent acquisitions across study visits. An automated advanced MRS acquisition protocol has been developed and suggested as an alternative to local MRS expertise [251]. With motion during the scan impairing data quality, imaging sites for HD clinical trials also benefit from previous experience of scanning participants with movement disorders.
Point Resolved Spectroscopy (PRESS), the most widely used MRS sequence in prior HD clinical research, is prone to localization errors due to refocusing pulses, which affect the appearance and quality of the obtained spectra. To mitigate localization errors, the more recently developed semi-LASER (sLASER) sequence makes use of semi-adiabatic localization by adiabatic selective refocusing, and yet achieves echo times close to those of PRESS. According to a consensus paper [252], the use of sLASER is one of the three most important factors for the successful application of MRS in the clinical setting (the other two being the use of simulated metabolite basis sets in analysis protocols and achieving a highly homogenous static magnetic field in the acquisition region).
Harmonization of data acquisition is critical for multi-site applications of MRS. Careful consideration should also be given to differences in acquisition and data quality between scanner makes and models, as those can compromise comparability of measurements for participants from different study sites. A recent paper [253] reported on an across-manufacturers (Philips, Siemens, GE) standardization of a short-echo sLASER sequence at 3T and reported consistent acquisition of high-quality spectra in regions including the putamen, with expectations of high reproducibility. Scanner change monitoring and management should also be included in longitudinal studies of cerebral metabolism, as scanner changes can occur over the follow up.
The output from the data acquisition voxel (volume of interest) is the MR spectrum, which graphically displays the detected signals as a function of their temporal frequencies. The linear combination model (LCModel) [254, 255] has been chosen by many studies for spectral fitting/metabolite quantification. It uses a non-linear least-squares algorithm to decompose spectra (temporal) in the frequency domain and directly simulates macro-molecule contributions, but not complete basis sets. A basis set acts to constrain the iterative peak fitting calculations for quantifying signal intensity values for the metabolites. The use of simulated metabolite basis sets could lead to improved spectral fitting with short echo times [252]. Several other models have been suggested and are being used alongside the LCModel (see e.g., https://mrshub.netlify.app/software_all/).
As with structural MRI, careful consideration should be given to selection of the region-of-interest for MRS, particularly if the trial involves surgical intracranial administration of the investigational medicinal product.
MEG
There are several challenges of incorporating MEG into clinical trials in HD. First and foremost, there are limited publications of MEG studies in HD to evaluate its usefulness. Ongoing work, such as the CLEAR-HD study, will start to address this. However, there are also logistical barriers to including MEG in future trials, such as low availability and accessibility. It is estimated that ∼160 MEG laboratories exist world-wide, with only 10 in the United Kingdom. Explanations for this include the requirement of sufficient space, the cost of the equipment, as well as the need for a magnetically shielded room (see Fig. 1).
Nonetheless, a handful of multi-center MEG studies have been conducted since 2011. A two-center study, with centers from the United Kingdom and the United States, used the same MEG system to investigate the age-related changes in the gamma band response [256]. A strong negative correlation between gamma band frequency and age was found. A four-center study of AD and controls in the United States used MEG scanners of the same system at two sites to acquire resting state MEG. Data from the two scanners were pooled and analyzed at a single center. Results showed global slowing in the AD patient group [257]. In an international, four-center study of mild cognitive impairment (MCI), the resting-state MEG dataset from one center was used to obtain specific features of MCI, which was applied to two datasets from the other sites to distinguish MCI patients from controls [258]. The specific features identified from the testing dataset were consistently present in the MCI groups across the datasets. Of note, the centers used the same MEG system. Recently, a two-site dataset for AD has been set up, consisting of 324 individuals – patients with MCI as well as controls [259]. The dataset includes resting-state MEG obtained from two sites with the same MEG system. The authors have reported the outcome of one standard preprocessing method, classifying the patients on the basis of the MCI, but they emphasize that this may not be the best pipeline – and that the classification was just above chance (i.e., 50%). Notably, a protocol for a repeated measures observational study to identify markers of disease progression in prodromal AD has included MEG (https://bmjopen.bmj.com/content/9/3/e024498; https://doi.org/10.1136/bmjopen-2018-024498). The protocol lists eight recruitment centers across the United Kingdom. Results are not yet available; however, once published, they will be informative with respect to the feasibility of multi-site MEG studies.
Overall, available evidence regarding the use of MEG in HD is minimal but increasing. Early evidence from its application in AD and PD suggests that MEG may be able to contribute to future clinical trials; however, more work is required to replicate and extend initial findings, and to develop stage-specific MEG signatures for HD.
SECTION 6: MULTI-MODAL NEUROIMAGING TO AID INTERPRETATION OF TRIAL DATA
The LEGATO-HD study is an example of how multi-modal imaging can be incorporated into a large Phase 2 trial, in order to better understand the effects of the therapy on the brain [91, 219]. In this trial, all participants underwent volumetric MRI at baseline and week 52, to investigate changes in brain atrophy in the treated groups compared with placebo. A subset also underwent MRS at selected sites only, to investigate changes in markers of neuronal integrity. A further much smaller subset also underwent TSPO PET-CT at a single site, to investigate changes in neuroinflammation (microglial activation) [91]. In this study, the imaging endpoints were defined as follows (simplified from the clinical protocol: https://classic.clinicaltrials.gov/ProvidedDocs/16/NCT02215616/Prot_000.pdf).
Percent change in caudate volume (baseline to week 52) in the treated groups compared with placebo
Percent change in whole-brain volume (baseline to week 52) in the treated groups compared with placebo Percent change in white-matter volume (baseline to week 52) in the treated groups compared with placebo Absolute change in ventricular volume (baseline to week 52) in the treated groups compared with placebo
TSPO PET-CT sub-study: change in microglial activation state (baseline to week 52) MRS sub-study: change in putaminal and frontal white matter markers of neuronal integrity (NAA) and astrocytosis (MI) (baseline to week 52).
By incorporating a range of imaging modalities into the same trial, the investigators were able to understand how the drug impacted on regional and global atrophy, regional neuronal integrity, and neuroinflammation. Of course, to fully realize the benefits of multi-modal imaging, it is vital that the image analyses are completed in a timely manner to allow interpretation of the data as a whole and not in isolation. This trial only had two imaging time-points – baseline and week 52. The addition of a third imaging time-point would have enabled a more robust analysis of trajectories of change. The addition of DWI may have provided valuable additional exploratory endpoints, and an understanding of the effects of the therapy on the microstructural integrity of the brain.
The SIGNAL-HD study is another example of a large (n = 265 HD gene-carriers), multi-center Phase 2 study in HD which included MRI and FDG-PET [175]. The study did not meet its pre-specified coprimary efficacy endpoints but reported a number of positive outcomes in the imaging analyses, including significant slowing of caudate atrophy at month 17 in the early-manifest cohort and widespread increase in FDG-PET signal compared to baseline in the cortex, but not the striatum. The use of validated volumetric MRI biomarkers like the caudate, provides strong evidence regarding a potentially beneficial effect of the drug on the brain, however volumetric measurements can be affected by other treatment-related effects, as discussed above, so it is important to relate to clinical outcomes and other biomarkers. The inclusion of FDG-PET in this study provided additional information regarding the mechanism of action of the treatment (e.g., a possible effect of pepinemab on astrocytes) and identified positive changes in many cortical regions, but not the striatum. The discordance between the volumetric and FDG-PET results in the striatum is important to note and highlights a potential issue with using multiple modalities, i.e., that results from different modalities do not always align and can sometimes even be contradictory. Preclinical imaging and information from natural history studies can help clarify the relationship between modalities, but there is a lot more work needed in this field to be able to bridge the gap.
Combining MR imaging – a note on cost
Volumetric MRI, DWI MRI, fMRI, and MRS are all MR techniques, which can be obtained during the same scanning session. MRI is typically charged to trial sponsors based on the total scan time and it would normally be possible to acquire all of the MR scans named above within a 1-hour timeframe. The addition of MRI with contrast, Computed Tomography (CT) or PET, to non-contrast MRI, would increase the total imaging cost substantially, but adding another type of non-contrast MR scan would not necessarily add to the total acquisition cost, unless that scan pushed the total scan time to over an hour. Although they are not widely available, the development of PET-MRI scanners has promise for the future in terms of reducing acquisition time and the need to travel between sites.
A note on statistical tests and statistical inference
Neuroimaging generates a vast amount of complex data which requires careful consideration of statistical inference methods for appropriate interpretation. Widespread use of statistical inference methods in scientific research has received scrutiny as outlined in the “ASA Statement on Statistical Significance and P-values” [260]. One important critique is that initial adoption of null hypothesis testing that was supposed to protect researchers from over-interpreting noisy data now appears to lead to the opposite effect. Others have elaborated on this, focusing on limitations of neuroimaging analyses methods- along with possible remedies [261]. One example is the minimum sample size (>12, or >20, or even more) that may be needed to draw scientifically robust conclusions. Another issue is the common use of parametric models used to capture underlying structure in data, which is thought to be explained by a fixed number of model parameters. For instance, many parametric models with Gaussian assumptions are applied regardless of the underlying data distribution [262].
Solutions to these issues may lie in being sufficiently adept in understanding statistical concepts before embarking on complex studies, developing novel generative statistical models using deep neural networks, more widespread use of non-parametric models, as well as with our willingness to use Bayesian approaches more often in our analyses of imaging datasets. Post-hoc analyses should always be interpreted with caution. Results based on small sample sizes should likewise be interpreted cautiously until replicated in a larger sample. Defining pre-processing and analytical pipelines a priori is critical, as is describing the reasons for such choices. The most important asset one might have in this regard is the awareness of limitations that may be inherent and acknowledging them in publications of results.
CONCLUSION
Neuroimaging is uniquely positioned to facilitate effective clinical trials in HD, across all the domains we have examined, from participant selection through to demonstration of disease modification (Table 2). To realize its potential, it is essential that careful consideration goes into the choice of imaging modality, outcome measure, analysis software and quality control. Careful site set-up and study design are also vital for obtaining sensitive and reliable data.
There are a number of recent developments which are likely to advance the utility of imaging in clinical trials in the future. For example, the identification of a mHTT PET ligand would allow direct imaging of the toxic agent in HD and measurements of the effect of mHTT-lowering therapies. Advances in hardware such as the integrated PET-MRI machines, collaborative efforts to harmonize imaging data acquisition and analysis, and the continual push to reduce acquisition times will improve practicability and data quality and reduce patient burden. These developments will need careful validation before widespread adoption but nevertheless are likely to lead to an increasingly prominent role of imaging in HD clinical trials.
Footnotes
ACKNOWLEDGMENTS
The authors acknowledge support from the European Huntington’s Disease Network (EHDN) in facilitating Imaging Working Group meetings. The authors would also like to acknowledge valuable input on the content of the article from EHDN Imaging Working Group members, particularly Claudia Metzler-Baddeley (Cardiff University), Carlos Estevez Fraga (UCL), Jan Kassubek (University of Ulm) and Hans-Peter Muller (University of Ulm).
FUNDING
RIS and NZH are funded by the Wellcome Trust, grant 223082/Z/21/Z.
MN is funded by the Huntington’s Disease Society of America for a pre-doctorate clinical fellowship.
CONFLICT OF INTEREST
MP and KM are employees of IXICO plc. The authors have no other conflicts of interest to report.