
Abstract
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder. The disease, characterized by motor, cognitive, and psychiatric impairments, is caused by the expansion of a CAG repeat in the huntingtin gene. Despite the discovery of the mutation in 1993, no disease-modifying treatments are yet available. Understanding the molecular and cellular mechanisms involved in HD is therefore crucial for the development of novel treatments. Emerging research has found that HD might be classified as a secondary tauopathy, with the presence of tau insoluble aggregates in late HD. Increased total tau protein levels have been observed in both HD patients and animal models of HD. Tau hyperphosphorylation, the main feature of tau pathology, has also been investigated and our own published results suggest that the protein phosphorylation machinery is dysregulated in the early stages of HD in R6/1 transgenic mice, primarily in the cortex and striatum. Protein phosphorylation, catalysed by kinases, regulates numerous cellular mechanisms and has been shown to be dysregulated in other neurodegenerative disorders, including Alzheimer’s disease. While it is still unclear how the mutation in the huntingtin gene leads to tau dysregulation in HD, several hypotheses have been explored. Evidence suggests that the mutant huntingtin does not directly interact with tau, but instead interacts with tau kinases, phosphatases, and proteins involved in tau alternative splicing, which could result in tau dysregulation as observed in HD. Altogether, there is increasing evidence that tau is undergoing pathological changes in HD and may be a good therapeutic target.
GENERAL INTRODUCTION ON TAU
Tau, Tubulin Associated Unit, is a microtubule-associated protein encoded by the MAPT gene located on human chromosome 17 (position 17q21). Tau is a soluble protein, predominantly expressed in neurons, composed of at most 441 amino acids (longest human isoform). Tau contains 4 functional domains: the N-terminal projection domain, the proline-rich region, the microtubule-binding domain, and the C-terminal region (Fig. 1). The microtubule-binding domain consists of four microtubule binding repeats (longest human isoform) that allow tau to regulate axonal transport by stabilizing microtubules and promoting tubulin assembly into microtubules. In addition to its microtubule-stabilizing functions, tau is thought to protect neuronal DNA [1] and regulate neurogenesis [2], neuronal activity [3], and long-term depression [4]. All these functions of tau have been highlighted in tau knock-out mice.
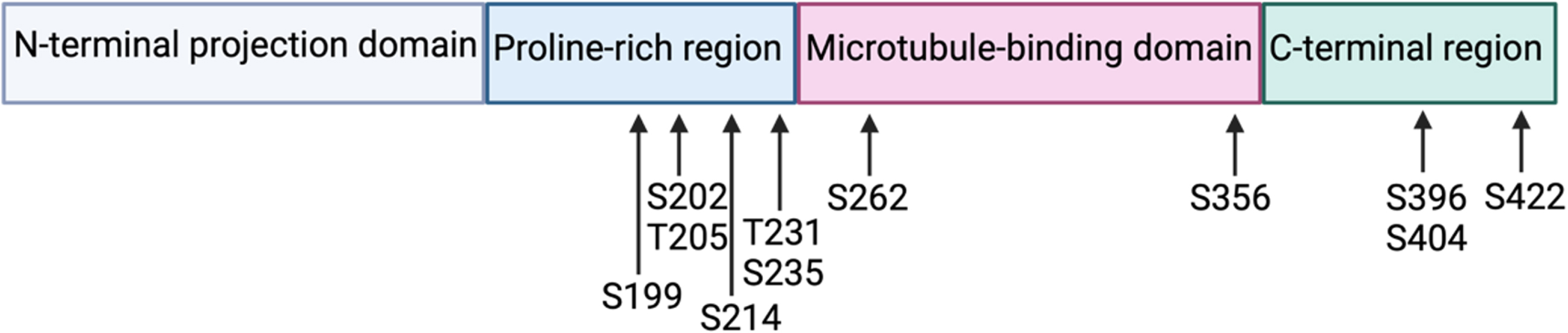
Tau protein with its 4 functional domains and the localization of the most studied phosphorylation residues.
Six tau isoforms are present in human brain tissue and are the result of alternative splicing of exons 2, 3, and 10. Alternative splicing of exon 10, which encodes the second microtubule-binding repeat, generates tau isoforms with either 3 (exon 10-) or 4 (exon 10+) microtubule-binding domains (3R, 4R) [5]. With an additional microtubule-binding domain, the 4R tau isoform has a stronger affinity for microtubules and is therefore more efficient for microtubule assembly [6]. The isoform ratio 4R/3R varies during development: foetal human brains express only the 3R tau isoform, while an equal amount of the 3R and 4R tau isoforms is found in adult human brains. In rodents, the 3R tau isoform is also expressed in foetal brains and adults express only the 4R tau isoform [7].
The alternative splicing of exons 2 and 3 leads to isoforms that differ in the presence of either 0, 1, or 2 29-amino-acid sequences in the N-terminal domain (0 N, 1 N, 2 N). The N-terminal domain has shown to regulate the neuronal subcellular localisation of tau, through interaction with non-microtubule proteins, such as membrane-binding proteins [8].
The MAPT gene is also characterized by 2 haplotype families consisting of a 900 kb chromosomal inversion (H1) and non-inversion (H2) [9]. Studies have shown that the MAPT haplotype can influence the development of neurodegenerative disorders: the H1 haplotype has been associated with progressive supranuclear palsy, corticobasal degeneration, and Alzheimer’s disease (AD) [10]. Interestingly, the H1 haplotype is also associated with greater levels of exon 10 containing transcripts (4R isoform) [11].
The importance of tau in neurodegeneration has been highlighted with the discovery of tau mutations implicated in the development of autosomal-dominant frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) [12]. Most of these mutations are known to affect exon 10 splicing, and thus the 4R/3R isoform ratio. This demonstrates that an imbalance in the 4R/3R isoform ratio is sufficient to provoke neurodegeneration associated with motor, cognitive and psychiatric disturbances. Moreover, it proves that a tau dysfunction can lead to neurodegeneration and is not only a consequence of neurodegenerative diseases.
Tau phosphorylation
Tau is post-translationally modified under physiological conditions, predominantly via phosphorylation: tau contains 85 potential phosphorylation residues (45 serine residues, 35 threonine residues, and 5 tyrosine residues) [13]. An increase in tau phosphorylation can modify the protein’s conformation and reduce its affinity for microtubules, thus providing tau with dynamic properties required for neuronal plasticity. More specifically, phosphorylation at the residues corresponding to the microtubule binding domains is imperative to regulate microtubule stabilization: phosphorylation at serine 262 and serine 356 is thought to induce the detachment of tau from microtubules [14] (Fig. 1.5). Phosphorylation at residues outside of the microtubule binding domains can also influence microtubule stabilisation, such as threonine 231 or serine 214 [13].
Under pathological conditions, tau is post-translationally modified to a higher degree. In tauopathies, tau hyperphosphorylation is thought to facilitate the aggregation of soluble tau filaments and the formation of insoluble aggregates made of twisted tau fibrils. Tau aggregates include neurofibrillary tangles (NFTs), present in AD, and round cytoplasmic inclusions (Pick bodies), typical of Pick’s disease [15]. As such, tau hyperphosphorylation seems to precede the formation of tau aggregates, but it is unclear if this process is sufficient for aggregate formation.
Specific phosphorylation sites have been associated with neurodegenerative disorders. For example, increased phosphorylation at serine 396 and serine 404 is implicated in the development of AD and Down syndrome [16]. Other phosphorylation sites found increased in NFTs include serine 214, serine 262, serine 356, and serine 422 [17].
However, the functional implications of tau hyperphosphorylation are difficult to analyse: the function of each phospho-site and the subsequent tau interactome remains unclear.
Mechanisms of tau phosphorylation
The transformation of tau into a phosphorylated state seems to be the result of a complex, coordinated, and sequential action of kinases and phosphatases [18]. Several kinases are involved in tau phosphorylation, including GSK-3β (glycogen synthase kinase 3 beta), CDK5, CaMKII, and PKA (protein kinase A) [9]. Protein phosphatases responsible for tau dephosphorylation include by PP1 (protein phosphatase 1), PP2A, PP2B, PP2C, PP3 and PP5. Among these, PP2A accounts for 70% of tau phosphatase activity in the human brain [19].
Tauopathies
Neurodegenerative disorders featuring hyperphosphorylated tau deposits are called tauopathies [20]. Tauopathies are clustered depending on the 4R/3R isoform ratio found in aggregates, the cell types in which the aggregates are found, the phosphorylation pattern, and the distribution in the brain. They are also divided into primary tauopathies, where tau is central to the disease, and secondary tauopathies, where tau aggregates are present alongside other protein aggregates.
TAU PATHOLOGY IN CLINICAL HUNTINGTON’S DISEASE
Emerging research has found that HD might be classified as a secondary tauopathy, with the presence of tau insoluble aggregates, altered tau levels, tau mis-splicing, tau hyperphosphorylation and tau truncation in HD brains [21, 22].
Tau aggregation
The presence of tau insoluble aggregates in HD patients was first highlighted in a case report in 1978, in which a 54-year-old HD patient showed NFTs in the entorhinal and parahippocampal cortex [23]. The first study involving several HD patients (n = 27) only came 20 years later, in which Jellinger and colleagues reported that 60% of HD patients showed tau pathology [24]. Following this, several clinical studies on postmortem HD brains showed the presence of aggregates composed of hyperphosphorylated tau in the cytoplasm and perinuclear space of cortical, striatal, and hippocampal neurons. These were evidenced using antibodies directed against phosphorylated tau, such as AT-8 (targeting phosphorylated tau at serine 202/threonine 205) [25, 26]. Other deposits, detected with antibodies that recognize 3R or 4R tau isoforms, have been found in the nucleus of cortical and striatal neurons. These rod-like deposits showed an ordered filamentous ultrastructure in the neuronal nuclear space of HD brains, and have thus been classified as “tau nuclear rods” (TNR) or “tau nuclear indentations” (TNI) [26, 27].
One study so far has evaluated tau accumulation in the brains of HD patients in vivo, using positron emission tomography (PET) imaging with the [18F]-AV-1451 tau tracer [28]. While this study included only 3 HD patients, results showed a pathological tau signal in all 3 patients, mainly in the striatum, cortex, and cerebellum. This study also showed that the most impaired HD patient displayed the highest uptake of tau tracer. In contrast, the least impaired patient had a weaker signal. While better powered studies are needed, this report is the first in vivo evidence of the implication of tau in HD patients.
Tau aggregates have also been found in early-onset HD patients (26 and 40 years at death) confirming that their presence is part of the disease mechanisms and unrelated to age [25]. However, a recent paper argues that tau pathology detected in HD most likely represents age-related changes instead of HD-related: they analyzed the regional distribution of tau and phospho-tau aggregation in 7 cases of HD, applying the newly defined criteria for tauopathies, with age-related neuronal (primarily age-related tauopathy, PART) and astroglial (aging-related tau astrogliopathy, ARTAG) tauopathy recognized as distinct entities [29]. Neuronal tau pathology was revealed in all cases. Five cases met the diagnostic criteria for PART, while one case was diagnosed with low levels of AD neuropathological changes (associated with subcortical amyloid deposition) and one case met the criteria for chronic traumatic encephalopathy (CTE, with characteristic perivascular phospho-tau). This study, although based on a small number of HD patients, extends previous tau studies in HD. However, these findings would have to be confirmed with a larger study.
Tau mis-splicing
In 2014, Fernandez-Nogales and colleagues highlighted an upregulated 4R/3R tau isoform ratio in the cortex and striatum of postmortem HD brains [26]. These results have since then been confirmed by other studies, reporting increased mRNA and protein levels of the 4R tau isoform in HD brains [25, 30]. These findings are of great importance since an imbalance in the 4R/3R tau isoform ratio can lead to neurodegeneration and could potentially participate in HD pathogenesis [31].
Tau hyperphosphorylation and expression levels
St-Amour et al., in 2018, were the first to quantify tau proteinopathy in HD human samples using a large cohort of individuals (n = 56). The study reports increased levels of tau phosphorylation in the later stages of HD, especially in Vonsattel grade 3 and 4 patients (grading scale for the severity of neuropathological involvement on postmortem HD brains, containing 5 grades, with grade 0 being unaffected and grade 4 being the most severe [32]). They were able to detect increased tau phosphorylation in the detergent-soluble fraction of the striatum at the serine 396, serine 404, serine 199, and threonine 205 sites. Tau oligomerization was also assessed by measuring tau phosphorylation in the sarkosyl-insoluble fraction, where increased levels of phosphorylation were observed at serine 396, serine 404, serine 199 and serine 235 [30]. Increased tau phosphorylation in HD brains has also been reported with immunohistochemical staining. Other studies have described the presence of neuronal inclusions in the striatum and cortex of HD patients, by using the AT-8 antibody, as explained previously [25, 26]. Recently, hyperphosphorylated tau was also detected in foetal neural grafts of HD patients, with the AT-8 and CP13 (targeting phosphorylated tau at serine 202) antibodies [33].
With regards to tau expression levels, Fernandez-Nogales et al. found increased tau protein levels in the cortex of HD brains [26]. In the striatum, another study reported a slight decrease in levels of soluble tau protein, and increased levels of tau mRNA [30].
Tau haplotype
The influence of the MAPT haplotype in the clinical expression of HD has also been investigated, as it is known that the haplotype regulates the expression of tau isoforms. Analysis of genotype/phenotype interaction on 960 HD subjects showed that HD patients carrying the H2 haplotype have a higher rate of cognitive decline compared to HD patients homozygous for the H1 haplotype [25]. This is particularly interesting, considering that the H1 haplotype has been associated with the development of neurodegenerative diseases, including progressive supranuclear palsy, corticobasal degeneration, and AD [10]. Moreover, the H2 haplotype has been considered as a protective haplotype in most tauopathies [10]. The mechanisms underlying the effect of the MAPT haplotype on cognitive decline in HD remain to be investigated.
Tau truncation
A soluble truncated tau species (Δtau314) was recently found elevated in the striatum (caudate nucleus) and prefrontal cortex of HD patients [34]. This increased amount of truncated tau has been correlated with an increased expression of caspase-2, which is also involved in the proteolysis of the mutant huntingtin [35]. Caspase-2 mediated tau cleavage on aspartate 314 is thought to be responsible for synaptic dysfunction and cognitive impairments in cellular and mouse models of frontotemporal dementia. The levels of this truncated species have also been shown elevated in AD patients and individuals with mild cognitive impairment. It suggests that this product is a common pathological mechanism that leads to synaptotoxicity in several neurodegenerative disorders [36]. However, these results indicate a defect in capsase-2 activity in HD, rather than a potential tau toxicity.
Tau in the cerebrospinal fluid
Total tau concentrations are also higher in the cerebrospinal fluid (CSF) of patients suffering from HD compared to healthy controls [37]. However, it does not specifically indicate that tau is involved in HD pathological mechanisms, as tau is thought to be released into the CSF after neuronal cell death [38]. Filipe Brogueira Rodrigues and colleagues investigated the suitability of CSF tau levels as a biomarker in HD. They found that the levels of tau in the CSF correlate with the phenotype severity in HD patients, as it predicted motor, cognitive, and functional impairment [39]. Consequently, this measurement could potentially be used in the future as a biomarker for disease progression and to evaluate the outcome of novel treatments.
TAU PATHOLOGY IN PRECLINICAL HUNTINGTON’S DISEASE MODELS
To further investigate tau involvement in HD, researchers have studied tau pathology in animal models as well [21].
Tau expression levels and mis-splicing
Fernandez-Nogales and colleagues found increased total tau protein levels in the striatum and cortex of 14-week-old R6/1 mice and in the cortex of 20-month-old HD94 mice (both at the fully symptomatic stage). They also found evidence of splicing dysregulation, as R6/1 mice presented higher levels of 4R tau protein in the striatum, and HD94 mice had a higher 4R/3R tau isoform ratio in both the striatum and cortex [26].
Tau hyperphosphorylation
Tau hyperphosphorylation, the main feature of tau pathology, has been investigated in HD mouse models, including the R6/2 mouse model [40, 41]. These mice exhibit a slight increase at the PHF1 epitope (serine 404/serine 396) in the hippocampus at the presymptomatic stage (3-week-old), expanding to the striatum and cortex at the fully symptomatic stage (10-week-old) [40, 41]. Fully symptomatic R6/2 mice also show an upregulation of the AT-8 epitope in the hippocampus, while total tau expression remained unchanged. Tau hyperphosphorylation was also examined in the Q175, a knock-in mouse model with a slower progression of the disease. Surprisingly, fully symptomatic Q175 mice showed unchanged levels at the PHF1 epitope, but showed increased phosphorylation at the serine 202 and serine 199 sites in the cortex [40]. The knock-in HD mouse model KI140 also showed an increase of phosphorylation at serine 396 from 17 months (fully symptomatic) of age in the cortex [41]. All these studies show a dysregulation of tau phosphorylation in HD mouse models. Some sites (serine 396) seem to be hyperphosphorylated even before the appearance of symptoms in these mice, suggesting that tau pathology may participate in the observed HD phenotype.
Tau aggregation
Tau nuclear inclusions have been found in the brains of fully symptomatic R6/1 and HD94 mouse models, by using antibodies that recognize either 4R tau isoform or total tau. However, these were less abundant in mouse brains compared to human brains [26].
The use of antibodies targeting tau phosphorylation has failed to detect tau aggregates in HD mouse models: studies on the R6/2 and KI140 mouse models reported the absence of aggregates using the phospho-serine 396 antibody [40, 41]. This absence contradicts clinical studies showing tau aggregates in postmortem HD brains, using antibodies targeting tau phosphorylation. These discrepancies can be explained by the fact that endogenous mouse tau is less prone to aggregation, compared to human tau. Furthermore, the phospho-epitopes of tau differ in different diseases and phosphorylation of serine 396 is known to occur at a later stage of disease in AD [42]. Using antibodies that specifically detect tau oligomers, such as the oligomeric antibody T22, may overcome the limitations of antibodies targeting phosphorylated tau [43].
Mechanisms leading to tau hyperphosphorylation in Huntington’s disease
It is still unclear how the mutation in the huntingtin gene leads to tau dysregulation in HD. One study has demonstrated that mutant huntingtin and tau deposits appear in the same subset of neurons. However, the two proteins do not interact, evidenced by the fact that they do not co-immunoprecipitate [26]. Therefore, several hypotheses have been explored, including the interaction of the mutant huntingtin with tau kinases and phosphatases (Fig. 1.6).
Tau kinases and phosphatases
To elucidate the mechanisms leading to tau hyperphosphorylation in HD, several tau kinases and phosphatases have been investigated in HD mouse models. Gratuze and colleagues investigated the expression and activation of several kinases in the hippocampus of fully symptomatic R6/2 mice. They found a decreased expression of CaMKII and a decreased activation of ERK1/2 [40]. The expression of CDK5 was also found downregulated in the cortex of fully symptomatic R6/2 and KI140 mice [41]. A decreased activity of GSK-3β has been found in the striatum of 8-week-old R6/1 mice (marked by an increased phosphorylation of GSK-3β at serine 9), followed by a decreased expression of the kinase by 14 weeks of age [44]. In the fully symptomatic R6/2 mice, there seems to be a decreased activity in the cortex but no changes in its expression [41]. Contrarily, another study showed that the expression of GSK-3β and its phosphorylated form (phosphorylated at tyrosine 216, active form) are increased in the hippocampus of postmortem HD brains [45]. These results were confirmed in the R6/2 mouse model, which show an increased phosphorylation at tyrosine 216 in the pre-symptomatic and symptomatic mice. The discrepancies seen in these studies might come from the fact that GSK-3β is regulated in vivo by mechanisms other than its phosphorylation at serine 9, which therefore is not an accurate measurement of its activity [46]. There is still no clear consensus about the activity and expression of this kinase in HD, and its involvement in tau hyperphosphorylation still needs to be defined.
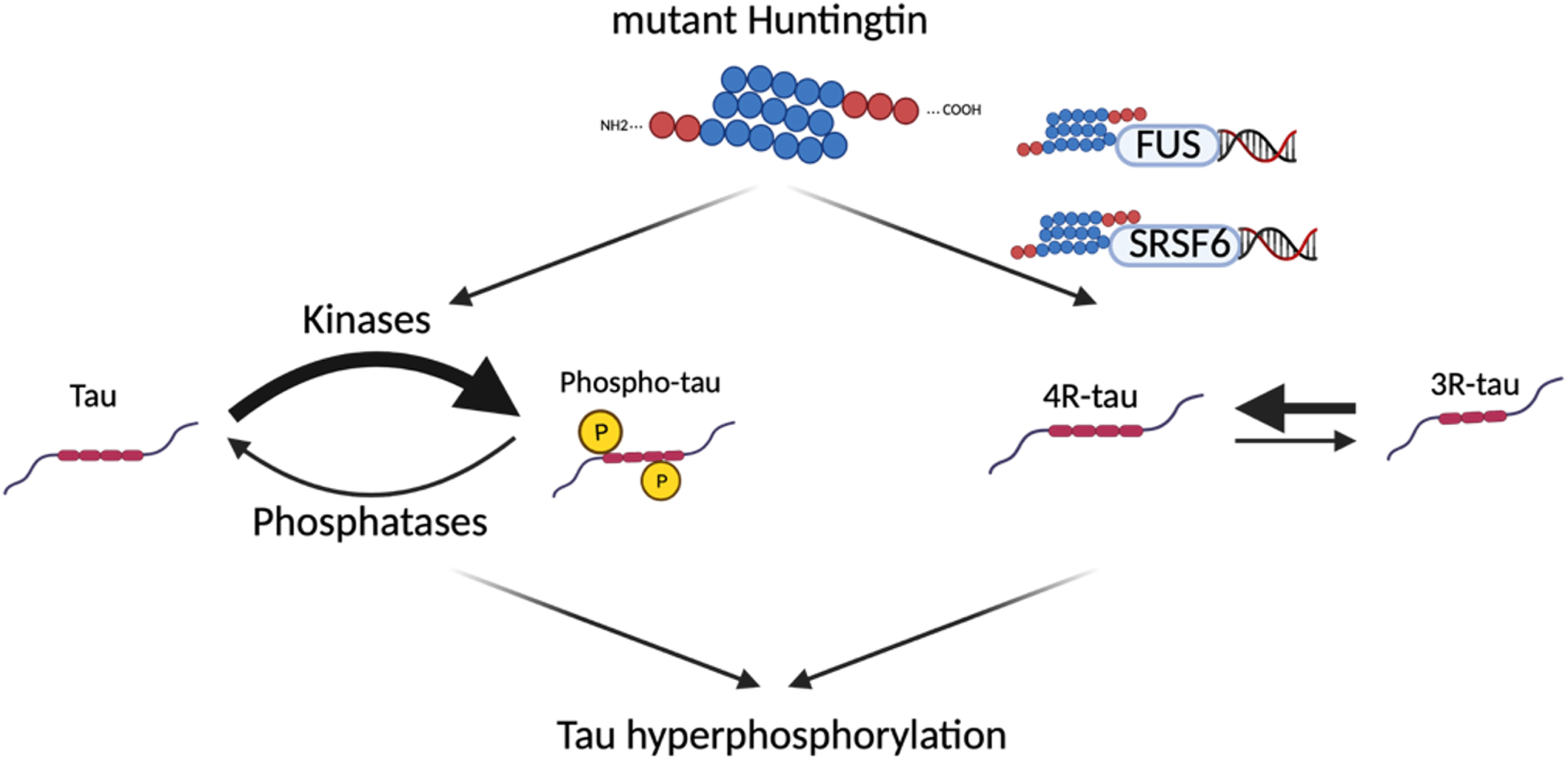
Potential mechanisms leading to tau hyperphosphorylation in Huntington’s disease. Since mutant huntingtin does not directly interact with tau, several hypotheses have been explored to explain the tau hyperphosphorylation seen in HD, including an interaction between the mutant huntingtin and tau kinases/phosphatases, and an interaction between mutant huntingtin and proteins involved in tau alternative splicing (FUS, SRSF6).
In summary, none of these studies report an increased expression or activity of tau kinases that would correlate with the exacerbated phosphorylation of tau.
When investigating the expression of tau phosphatases, researchers found that calcineurin (PP2B) is downregulated in the hippocampus of presymptomatic R6/2 mice (3 weeks of age) [40] and in the cortex of fully symptomatic R6/2 mice (10 weeks of age) [41]. Moreover, its decreased expression seems to be correlated with the increased tau phosphorylation (R2 = 0.5879). PP2B expression is also decreased the cortex of 12-month-old Q175 mice [40] and in the cortex of 17-month-old KI140 mice [41]. A reduction of calcineurin expression had previously been shown in HD patients [47]. These results indicate that a calcineurin-reduced expression could potentially be responsible for the hyperphosphorylation of tau in HD.
From mutant huntingtin to tau mis-splicing
Another potential indirect mechanism linking the mutant huntingtin and tau alteration has been found using bioinformatic tools [48]. A tau splicing factor, the serine/arginine-rich splicing factor 6 (SRSF6, also known as SRp55), has a 9 nucleotide consensus from which the last 8 correspond perfectly to the huntingtin CAG repeat sequence, and seems to bind to the mutant huntingtin mRNA in vitro [49]. SRSF6 splicing factor is known to promote tau exon 10 inclusion (4R tau isoform) [50]. Consequently, studies on brain samples from HD patients and the R6/1 mouse model have shown an accumulation of the SRSF6 in the huntingtin inclusions, and an increase in the total and phosphorylated forms of SRSF6. Moreover, co-transfection of SH-SY5Y neuroblastoma cells with SRSF6 and the stretched polyQ huntingtin resulted in an increase in the 4R/3R isoform ratio of tau mRNA [26].
An alternative mechanism has been proposed by Baskota et al. [29], involving the interaction between the DNA/RNA binding fused in sarcoma protein (FUS) and mutant huntingtin. The FUS protein has been associated with tau alternative splicing, as studies have identified an increased inclusion of tau exon 10 (tau 4R isoform) upon loss of FUS [51]. Interestingly, in vitro and in vivo studies on HD cellular and animal models found FUS to be a major component of mutant huntingtin aggregates [52, 53]. Moreover, it has been shown that co-aggregation between FUS and mutant huntingtin results in a depletion of free soluble FUS [53]. Therefore, the hypothesis is that the sequestration of FUS in huntingtin aggregates might be responsible for the altered tau isoforms observed in HD.
The involvement of these proteins and their interactions with mutant huntingtin still needs further investigation. Additionally, the mechanisms leading from tau mis-splicing to tau phosphorylation are still unknown.
Potential tau-driven toxicity in Huntington’s disease
Hyperphosphorylation of tau is known to prevent tau from binding to microtubules. In other pathologies, including AD, tau hyperphosphorylation weakens microtubule assembly and this function can be restored by enzymatic dephosphorylation [54]. Thus, we can, on the one hand, hypothesize that tau hyperphosphorylation can lead to a loss of function of the protein, by preventing its binding to microtubules. On the other hand, there is evidence that tau hyperphosphorylation, through a reduced affinity for microtubules, can facilitate its pathological cross-linking with other tau molecules and the formation of aggregates [55].
One of the most important microtubule-dependent functions of tau in neurons is its implication in retrograde and anterograde axonal transport, whose dysregulation can lead to cell death [56]. In the case of AD, neurons that exhibit NFTs show impaired anterograde transport in axons and impaired retrograde transport in dendrites [57]. Mis-localization and higher expression of tau are also known to modulate kinesin-1-dependent axonal transport [56, 58]. Kinesin-1 is a motor protein transporting cargoes into microtubules, including mitochondria and peroxisomes [59]. Accordingly, studies on HD cells and animal models have highlighted impaired mitochondrial transport in cortical and striatal neurons [60], but more studies are needed to investigate a connection to tau.
Impaired intraneuronal transport also involves the trafficking of neurotrophins. Increased expression, mislocalization, and hyperphosphorylation of tau have been associated with impaired neurotrophin signaling [61]. In the tauopathy mouse model expressing the 4R1N human tau isoform with the P301 S mutation, BDNF signaling was found to be impaired and BDNF TrkB receptors appeared uncoupled from their downstream signaling [61]. Decreased levels of BDNF mRNA and protein expression are found in the cortex and striatum of animal models of HD [62] and patients [63], along with a decreased expression of TrkB receptors [64]. Since striatal BDNF is mainly synthesized and transported anterogradely from other brain regions [65] (i.e. the cerebral cortex), abnormalities in axonal transport could be the cause for the lack of trophic support observed in HD. While mutant huntingtin is known to affect both retrograde and anterograde vesicle transport, the involvement of tau in these mechanisms has yet to be investigated in relation to HD.
Another potential tau-driven toxicity mechanism in HD comes from its involvement in the functions of T-complex protein 1-ring complex (TRiC). TRiC is a chaperonin protein, known to be neuroprotective in HD through the clearance of mutant huntingtin via axonal transport [66]. A study on WT neurons showed that TriC is also essential for BDNF retrograde axonal transport and that this function is dependent on tau [67]. It is still unclear whether the tau dysregulation observed in HD could exacerbate the lack of trophic support and the mutant huntingtin toxicity through its interaction with TRiC.
TAU TARGETED STRATEGIES FOR AD AND PRIMARY TAUOPATHIES
Given its role in disease, tau has been the target of several therapeutic strategies for the treatment of AD and primary tauopathies. These include small molecule inhibitors of tau post-translational modifications [68–70] (hyperphosphorylation, acetylation, glycosylation, truncation, etc.) and tau aggregation [71–74]. More recently, tau mRNA has been targeted using anti-sense oligonucleotides (ASO) [75–77]. Preclinical studies using this approach demonstrated reduced tau levels in the brain of tau transgenic mice, resulting in an overall reduction of tau pathology and protection against hippocampal volume loss, neuronal loss and nesting deficits [76]. A tau targeting ASO developed by Biogen and Ionis (BIIB080) is currently in Phase II clinical trial in patients with AD after being well tolerated in Phase I trials (clinical trial NCT05399888).
Immunotherapy is a promising pharmaceutical approach used to target tau, prevent its aggregation, and promote its clearance through microglial activation. There are currently two active tau immunotherapeutics in clinical trials for the treatment of AD. ACI-35 (clinical trial NCT04445831) is composed of a synthetic tau peptide phosphorylated at pathological residues S396 and S404 [78], whereas AADvac1 is composed of a non-phosphorylated synthetic tau peptide encompassing residues 294–305 [79] (clinical trial NCT02579252). As active immunotherapies activate both humoral and cellular immunity, however, it is difficult to manage any adverse effects. Passive immunotherapies on the other hand are deemed safer as they only induce humoral activity and currently dominate the AD clinical trial landscape. Monoclonal antibodies targeting tau at the amino terminus [80–83], the mid-domain [84, 85], phospho-epitopes [86–89], and oligomers [90–92] have been developed and characterized preclinically. Monoclonal antibodies targeting the amino terminal of tau have been clinically tested and although they were well tolerated, they failed to improve patient cognition [93, 94]. Monoclonal antibodies targeting the tau mid-domain (clinical trials NCT04658199, NCT04971733, NCT04619420), phospho-epitopes (clinical trial NCT04149860) and oligomers (clinical trial NCT05344989) are currently in Phase I and II clinical trials for the treatment of AD. The results from these studies may provide an insight into the best region of tau to target pharmaceutically.
THE SUITABILITY OF TAU AS A NEW THERAPEUTIC TARGET FOR HD
As previously mentioned, there is supporting evidence of tau being involved in the pathogenesis of HD, including our own study showing that tau is hyperphosphorylated at multiple sites in early symptomatic R6/1 HD mice [95]. However, although tau expression, phosphorylation, and conformation are thought to be altered in HD, there is limited evidence that tau affects the development of HD symptoms.
In a recent paper, we investigated the suitability of tau as a new therapeutic target for HD. To shine a light on the involvement of tau in HD, we aimed at 1) knocking out tau expression and 2) expressing a transgene encoding mutant human tau in the R6/1 mouse model of HD [96]. We hypothesized that expression of the mutant human tau transgene in HD mice would worsen the HD phenotype, while knocking out endogenous mouse tau in HD mice would improve some behavioral deficits displayed by HD mice. Our data suggested that neither the expression of a tau transgene nor the ablation of tau expression impacted the progression of the HD motor, cognitive and affective phenotypes. Our study contradicted a previously published study suggesting that tau contributes to the motor phenotype in HD, where R6/1 mice crossed with tau knockout mice exhibited milder motor impairments, compared to R6/1 mice expressing the tau protein.
While these discrepancies can possibly be explained by study differences (e.g., the use of different mouse strains), more studies are needed to evaluate and understand how tau influences the HD phenotype. Indeed, some of the limitations in the study we recently published include the aggressive progression of the HD phenotype in R6/1 mice, compared to the slow development of symptoms in Mapt-/- and P301 L mice. While Mapt-/- mice do not express tau during early development, these mice only show cognitive and motor impairments from 12 months of age [97, 98]. Furthermore, while P301 L mice express the transgene from the second postnatal week, motor and cognitive symptoms onset occurs at around 5 months of age [99, 100]. Therefore, crossbreeding HD mice with a milder disease progression with tau knockout mice and a tauopathy mouse model could potentially lead to different outcomes and would be crucial to validate these results.
Tau immunotherapy in HD mouse models
In a recent study, the role of hyperphosphorylated tau in HD and the potential of tau-targeted passive immunotherapy for the treatment of HD was investigated by treating zQ175 mice weekly with the anti-tau pS202 monoclonal antibody, CP13 [101]. After 12 weeks of treatment, hippocampal pS202 levels in the hippocampus were significantly reduced and mice displayed an improvement in motor behavior and cognitive health. Furthermore, CP13 treatment was demonstrated to reduce huntingtin oligomers; however, larger huntingtin aggregates were increased. This study therefore suggests that hyperphosphorylated tau may play a role in the behavioral and pathological hallmarks of HD and that targeting hyperphosphorylated tau may be a viable therapeutic strategy for the treatment of HD.
Targeting tau kinases in HD
Several tau kinases have been tested as potential targets to treat HD in preclinical models, including GSK-3β [102]. As previously mentioned, GSK-3β is a major kinase involved in tau phosphorylation [103]. Its involvement in HD pathogenesis has been investigated in several studies [104]. GSK-3β was found to accumulate with mutant huntingtin in neuronal lipid rafts, extracted from HD mice and HD primary neurons [105]. Furthermore, higher levels of GSK-3β expression (mRNA and protein) and activity have been found in the hippocampus of HD patients and R6/2 mice, correlating with an upregulation of tau phosphorylation at serine 202 and threonine 205 [45]. These studies highlighting a defective GSK-3β signaling in HD led to further studies investigating its suitability as a new therapeutic target.
In HD cell culture models, silencing GSK-3β and treatment with pharmacological inhibitors of GSK-3β have led to reduced levels of mutant huntingtin aggregates and neuronal death [105, 106]. Similar results have been obtained in HD mouse models, where treating R6/2 mice with a selective GSK-3 inhibitor led to improvements in motor coordination and neuroprotection [106]. Other studies have evaluated the effect of lithium, known to inhibit GSK-3β [107], in preclinical models of HD. A low-dose of lithium was demonstrated to have multiple beneficial effects on the YAC128 HD mouse model, including improvements in motor function and amelioration in the striatal neuropathological deficits [108]. Furthermore, co-treatment of lithium with valproate has shown additive beneficial effects in alleviating locomotor deficits and depressive-like behaviors in the YAC128 and N171-82Q HD mouse models [109].
CONCLUSIONS
Increasing evidence suggests that tau is undergoing pathological changes in HD and may be a good therapeutic target. However, as tau undergoes a high number of post-translational modifications, has multiple isoforms, and is cleaved to produce truncated peptides and aggregates, it is difficult to decide what exactly to target. Furthermore, pathological tau may look different depending on the disease. This means that an effective tau targeting drug for AD may not be successful for the treatment of HD. Furthermore, testing oligomer-specific antibodies or tau aggregation inhibitors may be futile if tau does not aggregate in HD or cannot be detected in HD transgenic models. Better characterization of pathological tau in HD is therefore paramount. In addition, knock-in mouse models expressing human tau may prove useful to better recapitulate human pathophysiology.
Footnotes
ACKNOWLEDGMENTS
The Florey Institute of Neuroscience and Mental Health acknowledges the support from the Victorian Government’s Operational Infrastructure Support Grant. AJH has been supported by a Principal Research Fellowship, Project Grants and an Ideas Grant from the National Health and Medical Research Council (NHMRC). TR currently holds a Ronald Philip Griffiths Fellowship and has been supported by an NHMRC Boosting Dementia Research Leadership Fellow. RMN has been supported by an NHMRC Ideas Grant and an Alzheimer’s Association Research Grant.
CONFLICT OF INTEREST
The authors have no competing interests to disclose.